Поговорим о том, как осуществляется нитрование толуола. Получают подобным взаимодействием огромное количество полуфабрикатов, используемых в изготовлении взрывчатых веществ, фармацевтических препаратов.
Значимость нитрования
Производные бензола в виде ароматических нитросоединений выпускаются в современной химической промышленности. Нитробензол является полупродуктом в анилинокрасочном, парфюмерном, фармацевтическом производстве. Он является отличным растворителем для многих органических соединений, включая и нитрит целлюлозы, формируя с ним желатинообразную массу. В нефтяной промышленности его применяют в качестве очистителя для смазочных масел. При нитровании толуола получают бензидин, анилин, фенилендиамин.
Характеристика нитрования
Нитрование характеризуется вводом группы NO2 в молекулу органического соединения. В зависимости от исходного вещества протекает данный процесс по радикальному, нуклеофильному, электрофильному механизму. В качестве активных частиц выступают катионы нитрония, ионы и радикалы NO2. Реакция нитрования толуола относится к замещению. Для других органических веществ возможно заместительное нитрование, а также присоединение по двойной связи.
Нитрование толуола в молекуле ароматического углеводорода осуществляется с помощью нитрующей смеси (серной и азотной кислот). Каталитические свойства проявляет выступающая в данном процессе в качестве водотнимающего средства.

Уравнение процесса
Нитрование толуола предполагает замещение одного водородного атома нитрогруппой. Как выглядит схема протекающего процесса?
Для того чтобы описать нитрование толуола, уравнение реакции можно представить в следующем виде:
ArH + HONO2+ = Ar-NO2 +H2 O
Оно позволяет судить только об общем ходе взаимодействия, но не раскрывает всех особенностей данного процесса. На самом деле происходит реакция между ароматическими углеводородами и продуктами азотной кислоты.
Учитывая, что в продуктах есть молекулы воды, это приводит к снижению концентрации азотной кислоты, поэтому нитрование толуола замедляется. Для того чтобы избежать подобной проблемы, осуществляют данный процесс при невысоких температурах, используя азотную кислоту в избыточном количестве.
Помимо серной кислоты, в качестве водоотнимающих средств применяют полифосфорные кислоты, трехфтористый бор. Они дают возможность снизать расход азотной кислоты, повышают эффективность взаимодействия.

Нюансы процесса
Нитрование толуола было описано в конце девятнадцатого века В. Марковниковым. Ему удалось установить связь между присутствием в реакционной смеси и скоростью протекания процесса. В современном производстве нитротолуола применяют безводную азотную кислоту, взятую в некотором избытке.
Кроме того, сульфирование и нитрование толуола связано с использованием доступного водоотнимающего компонента фторида бора. Его введение в реакционный процесс позволяет снижать стоимость получаемого продукта, что делает доступным нитрование толуола. Уравнение протекающего процесса в общем виде представлено ниже:
ArH + HNO3 + BF3= Ar-NO2 + BF3 ·H2 O
После завершения взаимодействия вводят воду, благодаря чему моногидрат фторида бора образует дигидрат. Его отгоняют в вакууме, затем добавляют фтористый кальций, возвращая соединение в исходный вид.

Специфика нитрования
Есть некоторые особенности данного процесса, связанные с выбором реагентов, субстракта реакции. Рассмотрим некоторые их варианты подробнее:
- 60-65 процентная азотная кислота в смеси с 96 процентной серной кислотой;
- смесь 98 % азотной кислоты и концентрированной серной кислот подходит для мало реакционных органических веществ;
- нитрат калия или аммония с концентрированной серной кислотой - это отличный выбор для производства полимерных нитросоединений.

Кинетика нитрования
Взаимодействующие со смесью серной и азотной кислот, нитруются по ионному механизму. В. Марковникову удалось охарактеризовать специфику данного взаимодействия. Процесс протекает в несколько стадий. Сначала образуется нитросерная кислота, которая подвергается диссоциации в водном растворе. Ионы нитрония вступают во взаимодействие с толуолом, образуя в качестве продукта нитротолуол. При добавлении в смесь молекул воды происходит замедление процесса.
В растворителях с органической природой - нитрометане, ацетонитриле, сульфолане - образование этого катиона позволяет увеличивать скорость нитрования.
Полученный катион нитрония прикрепляется к ядру ароматического толуола, при этом образуется промежуточное соединение. Далее происходит отрыв протона, приводящий к формированию нитротолуола.
Для детального описания происходящего процесса можно рассмотреть образование «сигма» и «пи» комплексов. Образование «сигма» комплекса является лимитирующей стадией взаимодействия. будет напрямую связана с быстротой присоединения катиона нитрония к атому углерода в ядре ароматического соединения. Отщепление протона от толуола осуществляется практически мгновенно.
Только в некоторых ситуациях могут возникать какие-то проблемы с замещением, связанные с существенным первичным кинетическим изотопным эффектом. Это связано с ускорением обратного процесса при наличии препятствий разного вида.
При выборе в качестве катализатора и водоотнимающего средства концентрированной серной кислоты наблюдается смещение равновесия процесса в сторону образования продуктов реакции.

Заключение
При нитровании толуола образуется нитротолуол, который является является ценным продуктом химической промышленности. Именно это вещество является взрывчатым соединением, поэтому востребовано на взрывных работах. Среди экологических проблем, связанных с его промышленным изготовлением, отметим использование существенного количества концентрированной серной кислоты.
Для того чтобы справиться с такой проблемой, химики ищут способы уменьшения сернокислотных отходов, получаемых после проведения процесса нитрования. Например, процесс осуществляют при пониженных температурах, применяют легко регенерируемые среды. Серная кислота обладает сильными окислительными свойствами, что негативно отражается на коррозии металлов, представляет повышенную опасность для живых организмов. При соблюдении всех норм безопасности можно справиться с этими проблемами, получать нитросоединения высокого качества.
Нитрование
Реакция нитрования является одной из наиболее изученных реакций ароматического замещения. Для препаративных целей нитрование, как правило, проводят смесью концентрированных азотной и серной кислот, так называемой нитрующей смесью . На первой стадии реакции происходит образование иона нитрония + NO 2 , который и является электрофильным агентом:
HO-NO 2 + H 2 SO 4 H 2 O + -NO 2 + HSO 4 -
H 2 O + -NO 2 + H 2 SO 4 H 3 O + + HSO 4 - + + NO 2
Наличие иона нитрония в этом растворе подтверждено спектроскопически. Азотная кислота в концентрированной серной кислоте практически нацело превращается в нитроний-катион. Незначительная эффективность самой азотной кислоты в реакции нитрования бензола объясняется низким содержанием иона + NO 2 .
B качестве нитрующих агентов используются также другие системы, в которых генерируется либо катион + NO 2 , либо соединение общей формулы NO2-Y где Y - хорошая уходящая группа. Некоторые из таких систем, нашедшие наибольшее применение, представлены в таблице 1 в порядке увеличения их активности.
Таблица 1. Нитрующие реагенты.
| Нитрующий реагент | Метод генерации | Арены, подвергающиеся нитрованию |
| Азотная кислота HO-NO2 |
Фенолы, эфиры фенолов, бифенил | |
| Ацетилнитрат CH 3 C(O)-O-NO 2 | СН 3 СООН + HNO 3 (СН 3 СО) 2 О + HNO 3 | Бензол, алкилбензолы |
| Диоксид азота N 2 O 4 (O=N-O-NO 2) |
Бензол, алкилбензолы | |
| Нитрующая смесь | H 2 SO 4 конц + HNO 3 | Бензол, алкилбензолы, галогенбензолы, бензойная кислота, нитробензол, нафталин |
| Хлорид нитрония Сl-NO 2 |
Бензол, алкилбензолы, нитробензол, | |
| Тетрафторборат нитрония BF 4 -+ NO 2 | HF . 2BF 3 + HNO 3 | Динитробензол |
На примере реакции нитрования алкилбензолов отчетливо прослеживается влияние пространственных факторов на направление электрофильного замещения. Так, при нитровании толуола (метилбензола) орто -изомер образуется в качестве основного продукта, а при переходе к этил-, изо -пропил- и особенно к трет -бутил-бензолу его выход существенно уменьшается (см. табл. 2).
Таблица 2. Влияние пространственых факторов на соотношение орто-, пара-изомеров в реакции нитрования (NO 2 +)
Замещение в положения, % |
|||
C 6 H 5 -C 2 H 5 |
|||
C 6 H 5 -CH(CH 3) 2 |
|||
C 6 H 5 -C(CH 3) 3 |
|||
При изучении нитрования алкилбензолов было обнаружено так называемое ипсо-замещение , когда электрофильная атака протекает по тому атому углерода бензольного кольца, которое уже содержит заместитель, например:

В отличие от нитрования, при галогенировании атака ароматического субстрата может осуществляться различными электрофилами. Свободные галогены, например, Cl 2 и Br 2 ,(прим.35) могут легко атаковать активированное ароматическое ядро (например, фенола), но не способны реагировать с бензолами и алкилбензолами (фотохимическая активация может, однако, в последнем случае привести к протеканию радикального замещения в боковую цепь; см. раздел IV.3). Для поляризации атакующей молекулы галогена необходим катализ кислотами Льюиса , такими как AlCl 3 , FeBr 3 , и т.п.; при этом в молекуле галогена появляется так называемый "электрофильный конец" (энергия же, требующаяся для образования катиона Наl + существенно выше). Тем самым электрофильное замещение существенно облегчается:

Галогенирование протекает очень энергично, если использовать реагенты, в которых галоген в результате поляризации имеет сильный положительный заряд или даже существует как катион. Так, очень инертный мета -динитробензол можно пробромировать бромом в концентрированной серной кислоте в присутствии сульфата серебра. Предполагают, что в этом случае промежуточно образуется бром-катион:
2Br 2 + Ag 2 SO 4 2Br + + 2AgBr + SO 4 2-
Реакционная способность элементарного иода в реакциях электрофильного замещения в ароматическом ядре незначительна, так что прямое иодирование возможно только в случае фенола и ароматических аминов. Иодирование других ароматических соединений проводят в присутствии окислителя (обычно, азотной кислоты). Считается, что в этих условиях роль электрофильного агента играет ион I- + OH 2 .

Для галогенирования аренов можно применять также смешанные галогены , например, монохлорид брома (BrCl) или иода (ICl):

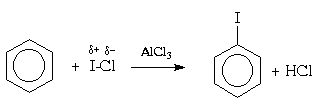
Галогенирование in vivo . В качестве примера электрофильного ароматического галогенирования, протекающего в живых организмах, можно привести реакцию иодирования -аминокислоты - тирозина в ходе биосинтеза иодсодержащих гормонов щитовидной железы до 3-иодтирозина и далее до 3,5-дииодтирозина:

Детали механизма сульфирования исследованы менее подробно по сравнению с нитрованием и галогенированием. Сам бензол сульфируется довольно медленно горячей концентрированной серной кислотой, но быстро - олеумом, SO 3 в инертных растворителях или комплексом SO 3 с пиридином. Природа электрофильной частицы зависит от условий реакции, но, вероятно, это всегда SO 3 , или в свободном состоянии, или связанный с "носителем", например, в виде H 2 SO 4 . SO 3 (H 2 S 2 O 7) в серной кислоте. Небольшие количества SO 3 образуются в H 2 SO 4:
2H 2 SO 4 SO 3 + H 3 O + + HSO 4 -
Атаку ароматического субстрата осуществляет атом серы поскольку он сильно положительно поляризован, то есть электронодефицитен:

Сульфирование является обратимым процессом. Это имеет практическое значение: при обработке сульфокислот водяным паромпроисходит замещение группы SO 3 Н на водород. Таким образом, можно ввести группу SO 3 Н как заместитель, ориентирующий требуемым образом последующие реакции (см. раздел IV.1.Б), а затем ее отщепить. Некоторые интересные особенности имеет сульфирование нафталина (см. раздел IV.1.Г).
Подобно галогенам, алкилгалогениды могут быть так сильно поляризованы кислотами Льюиса (хлоридами алюминия и цинка, трифторидом бора и др.), что они становятся способными к электрофильному замещению в ароматическом ядре:
R-Cl + AlCl 3 R +... Cl ...- AlCl 3 R + AlCl 4 -
Кроме алкилгалогенидов, источниками карбокатионов для галогенирования ароматических соединений могут быть алкены или спирты. При этом необходимо присутствие протонной кислоты, чтобы протонировать алкен или спирт. В случае спиртов требуется добавка не менее чем эквимольного количества кислоты (так как вода, выделяющаяся в ходе реакции, дезактивирует эквимольное количество катализатора), тогда как в реакциях с участием алкилгалогенидов и алкенов достаточно добавлять незначительное количество катализатора.
В лаборатории алкилирование по Фриделю-Крафтсу имеет ограниченное применение, так как обычно при этой реакции образуются смеси продуктов, что обусловлено рядом причин:
1) Образующийся продукт алкилирования легче вступает в реакции электрофильного ароматического замещения, чем исходное соединение (Alk - электронодонорная группа), поэтому дальше преимущественно алкилируется продукт. Если хотят получить продукты моноалкилирования, то необходимо брать большой избыток ароматического соединения.
2) Как и сульфирование, реакция алкилирования по Фриделю-Крафтсу обратима (см. также раздел IV.1.Г).
3) Даже в мягких условиях первичные и вторичные алкилгалогениды дают преимущественно вторичные или третичные алкиларены соответственно, поскольку алкилирование происходит в условиях, приближающихся к S N 1 реакции.(прим.37) Перегруппировки можно избежать, если работать при низких температурах.
Ацилирование ароматических соединений по Фриделю-Крафтсу является важнейшим методом синтеза жирноароматических кетонов. Производные карбоновых кислот, такие как ацилгалогениды и ангидриды, имеют полярную карбонильную группу и в принципе способны к электрофильному замещению в ароматических системах:

Электрофильная активность этих соединений, однако, невелика, и должна быть повышена действием кислот Льюиса. При этом кислотный катализатор, как правило, атакует атом кислорода карбонильного соединения и, смещая электронную плотность, повышает положительный заряд соседнего атома углерода. В результате образуется поляризованный комплекс (а в пределе - ацилкатион), действующий как электрофил:


Важное отличие реакции ацилирования ацилгалогенидами от реакции алкилирования алкилгалогенидами состоит в том, что в первой из этих реакций требуется более 1 моль кислоты Льюиса , тогда как во второй необходимо только каталитические количество. Это обусловлено тем, что кислота Льюиса образует комплекс как с ацилирующим производным карбоновой кислоты, так и с кетоном - продуктом реакции. При взаимодействии с ангидридами получающаяся кислота связывает еще моль моль катализатора, так что в целом его необходимо по крайней мере два моль. В каждом случае по окончании реакции образовавшийся комплекс кетона с хлоридом алюминия (или другой кислотой Льюиса) должен быть гидролитически разрушен (соляной кислотой со льдом).
Полиацилирования не наблюдается, поскольку образующийся кетон значительно менее реакционноспособен, чем исходное соединение (см. раздел IV.1.Б). Поэтому алкилбензолы часто предпочитают получать не прямым алкилированием, а ацилированием по Фриделю-Крафтсу с последующим восстановлением. Ароматические соединения с сильнодезактивирующими заместителями, например, нитро- или циано- группами, также не ацилируются по Фриделю-Крафтсу.
Контрольные задачи
2. Изобразите диаграмму потенциальной энергии
для реакции электрофильного ароматического
замещения, в которой медленной стадией является
образование
-комплекса (например,
нитрование бензола борфторидом нитрония;
см. раздел IV.1.A).
3. Какой продукт преимущственно образуется при бромировании: а)пара -нитротолуола; б) мета -нитробензолсульфокислоты; в) орто -нитрофенола.
4. Адреналин (1-(3",4"-дигидроксифенил)-2-метиламиноэтанол)- первый гормон, выделенный из мозгового вещества надпочечников, в настоящее время синтезируюют в три стадии из пирокатехина. Напишите уравнение первой стадии этого синтеза - реакции ацилирования пирокатехина (1,2-дигидроксибензола) хлорангидридом хлоруксусной киcлоты и объясните механизм).
5. Одной их качественных реакций на белки является ксантопротеиновая реакция, указывающая на присутствие ароматических -аминокислот. Она заключается в обработке белка азотной кислотой при нагревании. Напишите уравнение ксантопротеиновой реакции с тирозином (см. раздел I), образовавшимся в результате гидролиза белка.
Нитрование бензола может быть проведено с небольшими количествами исходных веществ, без выделения чистого продукта. Для получения нитробензола по уравнению:
С 6 Н 6 + HNO 3 à C 6 H 5 NO 2 + Н 2 О
необходима концентрированная азотная кислота (уд. вес. 1,4). Реакционная смесь при этом не должна нагреваться выше 50-60"С. При применении разбавленной кислоты реакция нитрования не идет; при повышении температуры начинается заметное образование динитробензола.
Из уравнения следует, что для реакции необходимы эквимолекулярные количества исходных веществ. Однако в таком случае реакция не дойдет до конца, так как выделяющаяся вода будет разбавлять азотную кислоту, и она потеряет нитрующее свойство. Следовательно, чтобы довести реакцию до конца, надо взять больше азотной кислоты, чем следует по теории. Но, чтобы реакция при этом не стала слишком бурной, азотную кислоту нужно растворить в концентрированной серной кислоте, которая не лишает азотную кислоту нитрующего действия и связывает выделяющуюся при реакции воду.
Чтобы предупредить возможность повышения температуры при реакции, не смешивают сразу все вещества, а к смеси кислот постепенно добавляют бензол. В небольшую колбочку наливают 8 мл концентрированной серной кислоты и 5 мл концентрированной азотной кислоты. Охлаждают смесь в струе воды. Затем к охлажденной смеси прибавляют небольшими порциями 4 мл бензола, постоянно встряхивая колбочку, чтобы достичь большего смешения нерастворяющихся друг в друге жидкостей (смесь кислот составляет нижний слой, бензол - верхний слой). После приливания всего бензола для достижения полноты реакции колбу закрывают пробкой с вертикальной трубкой (пары бензола летучи) и нагревают на предварительно нагретой до 60°С водяной бане
Время от времени колбу встряхивают, чтобы жидкости лучше перемешивались.
Продолжительность нагревания может определяться не столько необходимостью достижения полноты реакции, сколько наличием времени на уроке. При работе в кружке нагревание следует продолжать минут 30-40. Па уроке же удается продемонстрировать образование нитробензола после нагревания в течение 10 мин и даже вовсе без дополнительного нагревания, если реакция хорошо шла при приливании бензола к смеси кислот.
Нитробензол располагается слоем поверх смеси кислот. Выливают содержимое колбы в стакан с большим количеством воды. При этом кислоты растворяются в воде, нитробензол же собирается на дне стакана в виде тяжелой желтоватой жидкости. Если позволяет время, сливают часть жидкости с нитробензола и отделяют его с помощью делительной воронки.
При получении значительных количеств нитробензола и необходимости его очистки, нитробензол промывают водой, разбавленным (5-процентным) раствором щелочи, затем снова водой, разделяя всякий раз жидкости с помощью делительной воронки. После этого обезвоживают нитробензол, нагревая его с гранулированным хлоридом кальция, пока жидкость не станет прозрачной. Нагревание при этом необходимо, чтобы понизить вязкость нитробензола и достичь таким образом более полного контакта его с хлоридом кальция. Наконец, нитробензол может быть перегнан из небольшой колбочки с воздушным холодильником при температуре 204-207°С. Для того чтобы избежать разложения остатков динитробензола, не рекомендуется проводить перегонку досуха.
По физическим свойствам метилбензол (толуол) сходен с бензолом. При нормальных условиях он представляет собой бесцветную жидкость, нерастворимую в воде, но растворимую в органических растворителях. Подобно бензолу, он является хорошим растворителем органических соединений. В настоящее время толуол шире используют в качестве растворителя, чем бензол, из-за гораздо меньшей токсичности.
Химические свойства
Все реакции метилбензола можно подразделить на два типа: а) реакции, затрагивающие бензольное кольцо, и б) реакции, затрагивающие метильную группу.
Реакции в ароматическом кольце. Метилбензол вступает во все реакции электрофильного замещения, которые были описаны выше для бензола, а именно: в реакции нитрования, галогенирования, сульфирования и реакции Фриделя-Крафтса. Во всех этих реакциях метилбензол обнаруживает более высокую реакционную способность, и его реакции протекают с большей скоростью.
Нитрование метилбензола может осуществляться таким же способом, как и бензола. Продуктом нитрования метилбензола является смесь двух изомеров метилнитробензола:

Хлорирование толуола (метилбензола) в бензольное кольцо может проводиться путем пропускания через толуол газообразного хлора в присутствии хлорида алюминия (реакция проводится в темноте). Хлорид алюминия играет при этом роль катализатора. В этом случае образуются 2- и 4-замещенный изомеры:

Сульфирование метилбензола концентрированной серной кислотой тоже приводит к образованию смеси 2- и 4-замещенного изомеров:

Механизм всех этих реакций электрофильного замещения подобен механизму соответствующих реакций бензола. В этих реакциях 3-замещенные изомеры образуются в столь незначительных количествах, что ими можно пренебречь. Ниже эта особенность будет подробно обсуждена при рассмотрении направляющей (ориентирующей) способности замещающих групп.
Реакции в боковой цепи. Метильная группа в метилбензоле может вступать в определенные реакции, характерные для алканов, но также и в другие реакции, не характерные для алканов. Рассмотрим по одному примеру каждой из таких реакций.
Подобно алканам, метильная группа может галогенироваться по радикальному механизму. Для осуществления этой реакции хлор продувают через кипящий метилбензол в присутствии солнечного света или источника ультрафиолетового излучения. Отметим, что галогенирование бензольного кольца в метилбензоле требует совершенно иных условий.

Обратим внимание, что эта реакция представляет собой замещение. Дальнейшее галогенирование приводит к образованию следующих соединений:

Бромирование метилбензола осуществляется при аналогичных условиях и приводит к образованию соответствующих бромозамещениых соединений.
В предыдущем разделе указывалось, что алканы не вступают в реакции окисления даже с такими сильными окислителями, как перманганат калия. Однако метильная боковая цепь в метилбензоле подвергается окислению даже такими сравнительно мягкими окислителями, как оксид

Более сильные окислители, например перманганат калия, вызывают дальнейшее окисление:

Направляющее (ориентирующее) действие заместителей на бензольном кольце
Выше мы уже указывали, что электрофильное замещение метилбензола приводит к образованию 2- и 4-замещенного изомеров метилбензола. Поэтому метальную группу в метилбензоле называют 2,4-направляющей (или, иначе, ориентирующей в положения 2 и 4). Существуют и другие заместители на бензольном кольце, которые тоже обладают 2,4-направляющим действием в реакциях электрофильного замещения. Для таких реакций можно записать следующее общее уравнение:

В этом уравнении означает электрофил, а X-2,4-направляющий заместитель. Такие заместители обычно представляют собой насыщенные группы. К их числу относятся . При определенных условиях 2,4-направляющие заместители оказываются также 6-направляющими:

Реакции электрофильного замещения в положения 2 и 4 обычно имеют большую скорость, чем соответствующие реакции бензола.

К их числу относятся такие группы, как и . Реакции электрофильного замещения в положении 3 протекают медленнее, чем соответствующие реакции бензола.
Направляющая (ориентирующая) способность заместителей зависит от того, являются ли они донорами электронов для бензольного кольца или же, наоборот,
оттягивают от него электроны. 2,4-Замещение обусловливается положительным индуктивным эффектом либо положительным мезомерным эффектом. -Направляющие группы отдают электроны бензольному кольцу, вызывая указанные эффекты, и таким образом активируют кольцо. Поэтому они называются активирующими группами. 3-Направляющие группы оттягивают электроны от бензольного кольца, вызывая - и -эффекты. Они называются дезактивирующими группами.
Итак, повторим еще раз!
1. Ароматические соединения обладают следующими общими свойствами: а) горят с образованием дымного пламени;
(см. скан)
Рис. 18.7. Важнейшие реакции бензола.

Рис. 18.8. Важнейшие реакции метилбензола (толуола).
б) вступают в реакции замещения,
в) с трудом вступают в реакции присоединения.
2. Молекула бензола может рассматриваться как резонансный гибрид, образованный двумя предельными резонансными структурами.
3. Важнейшие химические реакции бензола указаны на рис. 18.7.
4. Важнейшие химические реакции метилбензола указаны на рис. 18.8.
5. В реакциях конденсации происходит соединение двух реагирующих молекул в новую молекулу с одновременным отщеплением небольшой молекулы какого-либо простого соединения, например воды или хлороводорода.
6. Насыщенные заместители на бензольном кольце обладают 2,4-направляющим (ориентирующим) действием.
7. Ненасыщенные заместители на бензольном кольце обладают З-иаправляю-щим (ориентирующим) действием.

Существование -комплексов, как устойчивых интермедиатов, Доказано прямыми опытами. Это не обязательно указывает на то, что переходное состояние реакции электрофильного присоединения - элиминирования сходно по структуре с -комплексом. В самом деле, вопрос о роли комплексов в этих реакциях все еще остается предметом дискуссии . В этом разделе основное внимание будет уделено механизму ароматического нитрования, который вызывает большой интерес . Затем будут рассмотрены Другие из перечисленных выше реакций электрофильного присоединения - элиминирования.
Результаты всех кинетических, спектроскопических и криоскопических работ, опубликованных до 1960 г., подтверждают точку зрения, что ион нитрония является эффективным электрофилом в реакциях нитрования аренов. С помощью спектроскопических и криоскопических методов были обнаружены низкие концентрации иона нитрония в безводной азотной кислоте. Надежно установлено также, что в присутствии большого избытка серной кислоты азотная кислота полностью превращается в нитронийбисульфат. Однако нитрование ароматических соединений, сходных по реакционной способности с бензолом, проходит в присутствии таких количеств воды, что ионы нитрония обнаружить невозможно. Вместе с тем участие ионов нитрония в нитровании также строго показано путем сравнения скорости нитрования со скоростью обмена 180 между средой и азотной кислотой. Реакции нитрования некоторых реакционноспособных субстратов в присутствии воды имеют нулевой кинетический порядок. Это указывает на то, что нитрующий агент образуется из азотной кислоты в медленной (лимитирующей) стадии, предшествующей атаке электрофила ароматическим кольцом. В отсутствие ароматического субстрата скорость обмена 180 между водой и азотной кислотой имеет нулевой порядок, как и при нитровании. Эти результаты лучше всего объясняются следующей схемой (уравнение 28):
Выделение нитрониевых солей, содержащих перхлорат-, тетра-фторборат- и гексафторфосфат-анионы, и тот факт, что эти соли являются эффективными нитрующими агентами, также свидетельствует в пользу ранее сделанных выводов.
Нитрование было использовано для установления относительной реакционной способности большого числа ароматических соединений. Относительные скорости замещения в и и -положения со статистическими поправками, так называемые парциальные факторы скорости, были рассчитаны для большого числа монозамещенных бензолов. Расчеты проводили, исходя из относительных скоростей по кинетическим данным или методом конкурентных реакций путем определения изомерного состава продукта реакции в тех же экспериментальных условиях. Очень близкие результаты были получены при нитровании эквимольных смесей бензола и толуола азотной кислотой в нитрометане, ацетонитриле, уксусном ангидриде или кислотных растворителях. Эти результаты также подтверждают универсальный характер механизма с участием нитроний-катиона. В табл. 2.5.2 приведены типичные примеры. Очевидно, что хотя распределение изомеров в реакциях с предварительно приготовленными нитрониевыми солями близко, к полученным ранее результатам, относительные скорости в реакциях с солями значительно ближе подходят друг к другу и, следовательно, парциальный фактор скорости, например для атаки метаположения, по-видимому, меньше, чем для отдельных положений бензола.
Таблица 2.5.2. Ориентация, относительная реакционная способность и парциальные факторы скорости при нитровании некоторых типичных молекул

Ниже приведен метод расчета на примере нитрования толуола сравнительно с бензолом в уксусной кислоте при . Напомним, что в бензоле имеется шесть эквивалентных положений, а в толуоле-одно пара-положение и по два орто- и мета-положений:

Изучение нитрования смесей бензола с толуолом с использованием предварительно приготовленных нитрониевых солей (см. табл. 2.5.2) и аналогичные эксперименты с м-ксилолом и мезитиленом, относительная реакционная способность которых по сравнению с бензолом оказалась равной 1,7 и 2,7 соответственно, увеличили число неясных вопросов. Выше уже упоминалось о непонятных результатах, полученных при расчете парциального фактора скорости для мета-положения. Кроме того, отмечалось, что в том случае, когда реакция успевает пройти в основном до того, как реагенты образуют достаточно однородную смесь, результаты определения реакционной способности из опытов по конкурентному замещению могут быть ошибочными.
Очевидно, что эта аномальная реакционная способность не подтверждается при определении констант скоростей нитрования нитрониевыми солями с помощью кинетических методов. Тем не менее были сделаны попытки обеспечить достаточное смешивание реагентов проведением реакции при различных их концентрациях. Эти результаты привели к предположению, что скорость ряда реакций аренов с электрофилами, в том числе и скорость нитрования нитрониевыми солями, связана скорее с -основностью, а не -основностью аренов. Иными словами, образование -комплекса является лимитирующей стадией. Однако место вхождения электрофила контролируется -основностью, как это показывают имеющиеся данные по распределению изомеров . Эта точка зрения на ароматическое нитрование была вновь подробно исследована . Было отмечено, что корреляция между соотношением продуктов нитрования нитрониевыми солями и относительной устойчивостью протонированных метилбензолов, по-видимому, ничуть не хуже, чем корреляция с устойчивостью -комплексов. Соотношение продуктов конкурентного нитрования полиметилбензолов смесью кислот в сульфолане также плохо коррелирует с устойчивостью -комплексов.
Вопрос, связанный со степенью прохождения реакции во время смешивания реагентов, изучен на примере нитрования бибензила, в котором каждое кольцо по реакционной способности аналогично толуолу, и, кроме того, передача влияния заместителя между кольцами минимальна. При использовании эквимольных концентраций бибензила и нитрониевой соли и полном смешении реагентов до начала реакции должны образоваться мононитробибензил (50%) и динитробибензил . С другой стороны, в случае неполного смешения реагентов до реакции должно увеличиться количество непрореагировавшего субстрата и динитробибеизила и уменьшиться количество мононитробибензила. Однако такое представление является слишком упрощенным, что со всей очевидностью следует из результатов нитрования бибензила азотной кислотой в уксусном ангидриде. Полнота смешивания не имеет значения для этой системы, но количество динитробибеизила составляет только 55% от ожидаемого. При нитровании бибензила в сульфолане нитронийтетрафторборатом с использованием различных концентраций и условий смешивания количество дизамещенного продукта всегда было значительно выше, чем это получается по приведенному расчету. Таким образом, простой метод конкурентных реакций не пригоден для определения относительной реакционной способности ароматических соединений при нитровании нитрониевыми солями. Очевидно, основное влияние оказывает скорость смешивания.
Интересные результаты были получены также при изучении кинетики нитрования аренов, реакционная способность которых выше, чем у бензола и толуола.
Можно было бы ожидать, что реакционная способность -ксилола, -ксилола и мезитилена значительно больше, чем у бензола (факторы около и ), однако при рассмотрении результатов нитрования в водной серной кислоте (68,3%) оказывается, что этот фактор ограничен значением 40. Предполагают, что лимитирующей стадией в этих реакциях является образование пары при столкновении между ионом нитрония и ароматическим субстратом. В чем различие между этим предположением и предположением о лимитирующем скорость образовании -комплексе? Ранее было отмечено, что соотношение продуктов плохо коррелирует с устойчивостью -комплексов. Эти факты можно объяснить, не прибегая к притягивающим взаимодействиям в пределах «пары столкновения». Энергетический барьер диссоциации «пары столкновения» на компоненты может превышать в кислой среде, этой энергии вполне достаточно для селективного образования -комплекса. Поскольку кинетические данные согласуются с диффузионной теорией соударений, то следует предпочесть термин "пара столкновения". Обсуждение всех приведенных выше данных приводит к заключению, что схему уравнений (28) следует видоизменить так, как это показано в уравнении (29), где определяет скорость только в исключительных обстоятельствах. Различные константы скорости атаки в орто-, мета- или пара-положения указывают, что распределение изомеров не зависит от лимитирующей стадии, поскольку в «пару столкновения» входит вся молекула арена, а не отдельные ее атомы.

Обсуждение нитрования можно удобно обобщить в виде энергетических профилей, как это сделано на рис. 2.5.1. Экспериментальные результаты показывают, что в случае нитрования бензола и толуола скорость определяющей стадией является образование -комплекса (рис. а и б соответственно), а образование «пары столкновения» определяет скорость нитрования псевдокумола (-триметилбензола) (рис. в).
Распределение изомеров, которое было обнаружено при нитровании толуола (см. табл. 2.5.2.), в основном не зависит от условий реакции, исключая обычное влияние температуры. Однако необычные результаты, полученные при изучении некоторых полиметилбензолов, осложняют понимание реакции нитрования . Так, нитрование азотной кислотой в уксусном ангидриде приводит к продуктам ацетоксилирования. Это смешивает все карты, заставляя предположить, что проходит электрофильное ацетоксилирование. В настоящее время известно, что это не имеет места.


Рис. 2.5.1. Профили энергии для реакций нитрования бензола (а) и толуола (б) и реакции образования 5- и : а, б-лимитирующей стадией является образование -комплекса; в - лимитирующей стадией является образование пары столкновения.
Хотя азотная кислота присутствует, главным образом, в виде ацетилнитрата, все же имеются некоторые сомнения относительно истинной природы электрофила. Так, азотная кислота в ускусном ангидриде реагирует с -ксилолом с образованием смеси -диметилнитробензола и -диметилфенилацетата. Выделение аддуктов, в которых ацетилнитрат присоединяется по замещенному положению (ипсо-атака), и тот факт, что эти аддукты разлагаются, очевидно, по внутримолекулярному механизму с образованием ацетата (57), свидетельствуют в пользу механизма, приведенного на схеме уравнений (30):

Аналогичные аддукты (58)-(64) выделены в реакциях с другими аренами, а некоторые аддукты были обнаружены в качестве интермедиатов. При реакции азотной кислоты в уксусном ангидриде с n-трет-бутилтолуолом при низкой температуре получены и выделены три аддукта. Основным продуктом является -аддукт (64), который, по-видимому, образуется в большем количестве, чем два других изомера, за счет пространственных затруднений в положении 4 (ипсо к трет-Ви). Другие нуклеофилы, например нитрат-ион и вода, также могут присоединяться к ипсо-замещенным -комплекса . Ипсо-Замещенные -комплексы можно регенерировать ацидолизом.

Нитрование о-ксилола в серной кислоте дает -диметилнитробензол и -диметилнитробензол, выход которых зависит от кислотности системы. При низкой кислотности получаются также нитродиметилфенолы. При ацидолизе эфиров (58) и (59) образуется -диметилнитробензол, выход которого увеличивается при возрастании кислотности; это подтверждает предположение, что нитрование ортоположения к алкильной группе, вероятно, часто включает первоначальную ипсо-атаку. Тот факт, что при ацидолизе (58) и (59) не образуется -диметилнитробензола, означает, что первоначальный ипсо-замещенный о-комплекс не превращается вновь в «пару столкновения» и что в нем не происходят ни , ни последовательные -сдвиги нитрогруппы. Эти результаты обобщены в уравнениях (31). Увеличение выхода и при возрастании кислотности среды при нитровании -триметилбензола в серной кислоте приписано миграции нитрогруппы от к и от к соответственно.

В настоящее время не известно ни одного достоверного примера последовательных внутримолекулярных -сдвигов через незамещенное положение. Однако известны последовательные -сдвиги из одного ипсо-положения через другое ипсо-положение. Например, при ацидолизе (65) образуются структурные изомеры (66) и (67), как показано в уравнении (32).

Раньше упоминалось об отщеплении ипсо-заместителя, например в случае протодесилилирования и протодесульфирования (см. с. 330,331). С этой точки зрения мы обсудим только замещение ипсо-заместителя на нитрогруппу. Многие реакции этого типа известны уже с давних пор, однако их механизм, как правило, не выяснен. К таким реакциям относятся дезалкилирование, дезацилирование, десилилирование, десульфирование, декарбоксилирование, дедиазотирование и дегалогенирование . Реакции с участием субстратов, сильно активированных по отношению к электрофильной атаке, могут включать первоначальное нитрозированце с последующим окислением.







