Parlons de la manière dont s'effectue la nitration du toluène. Grâce à une telle interaction, on obtient un grand nombre de produits semi-finis utilisés dans la fabrication d'explosifs et de produits pharmaceutiques.
Importance de la nitration
Les dérivés du benzène sous forme de composés nitrés aromatiques sont produits dans l'industrie chimique moderne. Le nitrobenzène est un produit intermédiaire dans la production de colorants aniline, de parfums et de produits pharmaceutiques. C'est un excellent solvant pour de nombreux composés organiques, dont le nitrite de cellulose, formant avec lui une masse gélatineuse. Dans l’industrie pétrolière, il est utilisé comme purificateur d’huiles lubrifiantes. La nitration du toluène produit de la benzidine, de l'aniline et de la phénylènediamine.
Caractéristiques de nitration
La nitration est caractérisée par l'introduction d'un groupe NO2 dans la molécule d'un composé organique. Selon la substance de départ, ce processus se déroule selon un mécanisme radicalaire, nucléophile ou électrophile. Les cations nitronium, les ions NO2 et les radicaux agissent comme des particules actives. La réaction de nitration du toluène est une réaction de substitution. Pour d'autres substances organiques, une nitration substitutive est possible, ainsi qu'une addition par double liaison.
La nitration du toluène dans une molécule d'hydrocarbure aromatique est réalisée à l'aide d'un mélange nitrant (acides sulfurique et nitrique). Il agit comme agent d’élimination de l’eau dans ce processus et présente des propriétés catalytiques.

Équation de processus
La nitration du toluène implique le remplacement d'un atome d'hydrogène par un groupe nitro. À quoi ressemble l’organigramme du processus ?
Pour décrire la nitration du toluène, l'équation de réaction peut être représentée comme suit :
ArH + HONO2+ = Ar-NO2 +H2O
Il permet de juger uniquement du déroulement général de l'interaction, mais ne révèle pas toutes les caractéristiques de ce processus. Ce qui se produit réellement est une réaction entre les hydrocarbures aromatiques et les produits acides nitriques.
Étant donné que les produits contiennent des molécules d'eau, cela entraîne une diminution de la concentration d'acide nitrique, ce qui ralentit la nitration du toluène. Afin d'éviter ce problème, ce procédé est réalisé à basse température, en utilisant de l'acide nitrique en quantité excessive.
Outre l'acide sulfurique, les acides polyphosphoriques et le trifluorure de bore sont utilisés comme agents déshydratants. Ils permettent de réduire la consommation d'acide nitrique et d'augmenter l'efficacité de l'interaction.

Nuances de processus
La nitration du toluène a été décrite à la fin du XIXe siècle par V. Markovnikov. Il a pu établir un lien entre la présence dans le mélange réactionnel et la vitesse du processus. Dans la production moderne de nitrotoluène, on utilise de l'acide nitrique anhydre, pris en excès.
De plus, la sulfonation et la nitration du toluène impliquent l'utilisation du fluorure de bore, un composant éliminant l'eau disponible. Son introduction dans le procédé réactionnel permet de réduire le coût du produit obtenu, ce qui rend accessible la nitration du toluène. L’équation du processus en cours est présentée sous forme générale ci-dessous :
ArH + HNO3 + BF3= Ar-NO2 + BF3 ·H2 O
Une fois la réaction terminée, de l'eau est introduite, grâce à quoi le fluorure de bore monohydraté forme un dihydrate. Il est distillé sous vide, puis du fluorure de calcium est ajouté, ramenant le composé à sa forme originale.

Spécificités de la nitration
Certaines caractéristiques de ce procédé sont liées au choix des réactifs et du substrat de réaction. Examinons certaines de leurs options plus en détail :
- 60 à 65 pour cent d'acide nitrique mélangé à 96 pour cent d'acide sulfurique ;
- un mélange d'acide nitrique à 98 % et d'acide sulfurique concentré convient aux substances organiques légèrement réactives ;
- Le nitrate de potassium ou d'ammonium avec de l'acide sulfurique concentré est un excellent choix pour la production de composés polymères nitro.

Cinétique de nitration
Réagissant avec un mélange d'acides sulfurique et nitrique, ils sont nitrés par un mécanisme ionique. V. Markovnikov a réussi à caractériser les spécificités de cette interaction. Le processus se déroule en plusieurs étapes. Tout d'abord, il se forme de l'acide nitrosulfurique, qui subit une dissociation dans une solution aqueuse. Les ions nitronium réagissent avec le toluène, formant du nitrotoluène comme produit. Lorsque des molécules d’eau sont ajoutées au mélange, le processus ralentit.
Dans les solvants à caractère organique - nitrométhane, acétonitrile, sulfolane - la formation de ce cation permet d'augmenter le taux de nitration.
Le cation nitronium résultant se fixe au noyau de toluène aromatique, formant un intermédiaire. Ensuite, l’abstraction de protons se produit, conduisant à la formation de nitrotoluène.
Pour une description détaillée du processus en cours, nous pouvons considérer la formation des complexes « sigma » et « pi ». La formation du complexe « sigma » est l’étape limitante de l’interaction. sera directement liée à la vitesse d'addition du cation nitronium à l'atome de carbone dans le noyau du composé aromatique. L’élimination d’un proton du toluène se produit presque instantanément.
Ce n'est que dans certaines situations qu'il peut y avoir des problèmes de substitution associés à un effet isotopique cinétique primaire significatif. Cela est dû à l'accélération du processus inverse en présence de divers types d'obstacles.
Lors du choix de l'acide sulfurique concentré comme catalyseur et agent d'élimination de l'eau, on observe un déplacement de l'équilibre du processus vers la formation de produits de réaction.

Conclusion
La nitration du toluène produit du nitrotoluène, un produit précieux de l'industrie chimique. Cette substance est un composé explosif, elle est donc très demandée lors des opérations de dynamitage. Parmi les problèmes environnementaux liés à sa production industrielle, on note l’utilisation d’une quantité importante d’acide sulfurique concentré.
Pour résoudre ce problème, les chimistes recherchent des moyens de réduire les déchets d’acide sulfurique produits après le processus de nitration. Par exemple, le processus est effectué à basse température et des milieux facilement régénérables sont utilisés. L'acide sulfurique possède de fortes propriétés oxydantes, ce qui affecte négativement la corrosion des métaux et présente un danger accru pour les organismes vivants. Si vous respectez toutes les normes de sécurité, vous pouvez surmonter ces problèmes et obtenir des composés nitro de haute qualité.
Nitration
La réaction de nitration est l’une des réactions de substitution aromatique les plus étudiées. À des fins préparatoires, la nitration est généralement effectuée avec un mélange d'acides nitrique et sulfurique concentrés, appelés mélange nitrant . Lors de la première étape de la réaction, il se produit la formation d'ions nitronium + NO 2, qui est un agent électrophile :
HO-NO 2 + H 2 SO 4 H 2 O + -NO 2 + HSO 4 -
H 2 O + -NO 2 + H 2 SO 4 H 3 O + + HSO 4 - + + NON 2
La présence d'ions nitronium dans cette solution a été confirmée par spectroscopie. L'acide nitrique présent dans l'acide sulfurique concentré est presque entièrement converti en cation nitronium. L'efficacité insignifiante de l'acide nitrique lui-même dans la réaction de nitration du benzène s'explique par la faible teneur en ion + NO 2.
D'autres systèmes sont également utilisés comme agents nitrants, dans lesquels un cation + NO 2 ou un composé de formule générale NO2-Y est généré dans lequel Y est un bon groupe partant. Certains de ces systèmes qui ont trouvé la plus grande application sont présentés dans le tableau 1 par ordre d'activité croissante.
Tableau 1. Réactifs nitrants.
| Réactif nitrant | Méthode de génération | Arènes soumises à la nitration |
| Acide nitrique HO-NO2 |
Phénols, éthers de phénol, biphényle | |
| Nitrate d'acétyle CH 3 C(O)-O-NO 2 | CH 3 COOH + HNO 3 (CH 3 CO) 2 O + HNO 3 | Benzène, alkylbenzènes |
| Dioxyde d'azote N 2 O 4 (O = N-O-NO 2) |
Benzène, alkylbenzènes | |
| Mélange nitrant | H 2 SO 4 concentré + HNO 3 | Benzène, alkylbenzènes, halobenzènes, acide benzoïque, nitrobenzène, naphtalène |
| Chlorure de nitronium Cl-NO 2 |
Benzène, alkylbenzènes, nitrobenzène, | |
| Tétrafluoroborate de nitronium BF 4 -+ NO 2 | H.F. 2BF 3 + HNO 3 | Dinitrobenzène |
En utilisant l'exemple de la réaction de nitration des alkylbenzènes, l'influence des facteurs spatiaux sur la direction de la substitution électrophile est clairement visible. Ainsi, lors de la nitration du toluène (méthylbenzène) ortho-l'isomère est formé comme produit principal et lors de la transition vers l'éthyle-, iso-bu- et surtout à frotte-butyl-benzène, son rendement diminue significativement (voir tableau 2).
Tableau 2. Influence des facteurs spatiaux sur le rapport des isomères ortho-, para dans la réaction de nitration (NO 2 +)
Remplacement dans les postes, % |
|||
C6H5-C2H5 |
|||
C6H5-CH(CH3)2 |
|||
C6H5-C(CH3)3 |
|||
Lors de l'étude de la nitration des alkylbenzènes, ce qu'on appelle ipso-substitution , lorsqu'une attaque électrophile se produit au niveau de l'atome de carbone du cycle benzénique qui contient déjà un substituant, par exemple :

Contrairement à la nitration, lors de l'halogénation l'attaque du substrat aromatique peut être réalisée par divers électrophiles. Les halogènes libres, par exemple Cl 2 et Br 2 (note 35), peuvent facilement attaquer le cycle aromatique activé (par exemple le phénol), mais ne sont pas capables de réagir avec les benzènes et les alkylbenzènes (l'activation photochimique peut cependant dans ce dernier cas conduire à l'apparition de substitutions radicales de chaînes latérales ; voir section IV.3). Pour polariser la molécule halogène attaquante, il faut Catalyse acide de Lewis tels que AlCl 3, FeBr 3, etc.; dans ce cas, ce que l'on appelle « l'extrémité électrophile » apparaît dans la molécule halogène (l'énergie nécessaire à la formation du cation Hal + est nettement plus élevée). Cela rend la substitution électrophile beaucoup plus facile :

L'halogénation se déroule très vigoureusement si l'on utilise des réactifs dans lesquels l'halogène, en raison de la polarisation, a une forte charge positive ou existe même sous forme de cation. Oui, très inerte méta Le -dinitrobenzène peut être bromé avec du brome dans de l'acide sulfurique concentré en présence de sulfate d'argent. On suppose que dans ce cas un cation brome se forme intermédiairement :
2Br 2 + Ag 2 SO 4 2Br + + 2AgBr + SO 4 2-
La réactivité de l'iode élémentaire dans les réactions de substitution électrophile dans le cycle aromatique est négligeable, donc l'iodation directe n'est possible que dans le cas du phénol et des amines aromatiques. L'iodation des autres composés aromatiques s'effectue en présence d'un agent oxydant (généralement de l'acide nitrique). On pense que dans ces conditions, le rôle d'agent électrophile est joué par l'ion I- + OH 2.

Pour l'halogénation des arènes, il peut également être utilisé halogènes mélangés, par exemple, le monochlorure de brome (BrCl) ou l'iode (ICl) :

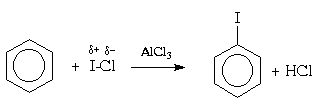
Halogénation in vivo. A titre d'exemple d'halogénation aromatique électrophile se produisant dans les organismes vivants, on peut citer la réaction d'iodation de l'acide aminé tyrosine lors de la biosynthèse des hormones thyroïdiennes iodées en 3-iodotyrosine puis en 3,5-diiodotyrosine :

Les détails du mécanisme de sulfonation ont été étudiés de manière moins détaillée que ceux de la nitration et de l'halogénation. Le benzène lui-même est sulfoné assez lentement avec de l'acide sulfurique concentré chaud, mais rapidement avec de l'oléum, du SO 3 dans des solvants inertes ou un complexe de SO 3 avec la pyridine. La nature de l'espèce électrophile dépend des conditions de réaction, mais il s'agit probablement toujours de SO 3 , soit à l'état libre, soit lié à un « support », par exemple sous forme de H 2 SO 4. SO 3 (H 2 S 2 O 7) dans l'acide sulfurique. De petites quantités de SO 3 se forment dans H 2 SO 4 :
2H 2 SO 4 SO 3 + H 3 O + + HSO 4 -
L’attaque du substrat aromatique est réalisée par l’atome de soufre puisqu’il est fortement polarisé positivement, c’est-à-dire déficient en électrons :

La sulfonation est réversible processus. Ceci est d'une importance pratique : lorsque les acides sulfoniques sont traités à la vapeur, le groupe SO 3 H est remplacé par de l'hydrogène. Ainsi, il est possible d'introduire le groupe SO 3 H comme substituant qui oriente les réactions ultérieures de la manière requise (voir section IV.1.B), puis de le supprimer. La sulfonation du naphtalène présente des caractéristiques intéressantes (voir section IV.1.D).
Comme les halogènes, les halogénures d'alkyle peuvent être très polarisés Acides de Lewis(chlorures d'aluminium et de zinc, trifluorure de bore, etc.) qu'ils deviennent capables de substitution électrophile dans le cycle aromatique :
R-Cl + AlCl 3 R +... Cl ...- AlCl 3 R + AlCl 4 -
Outre les halogénures d'alkyle, les alcènes ou les alcools peuvent être des sources de carbocations pour l'halogénation des composés aromatiques. Dans ce cas, la présence d’un acide protique est nécessaire pour protoner l’alcène ou l’alcool. Dans le cas des alcools, un additif est requis pas moins qu'équimolaire quantité d'acide (puisque l'eau libérée pendant la réaction désactive une quantité équimolaire du catalyseur), alors que dans les réactions impliquant des halogénures d'alkyle et des alcènes, il suffit d'ajouter une petite quantité de catalyseur.
En laboratoire, l'alkylation de Friedel-Crafts a une utilisation limitée, car cette réaction produit généralement des mélanges de produits, pour plusieurs raisons :
1) Le produit d'alkylation résultant subit plus facilement des réactions de substitution aromatique électrophile que le composé de départ (Alk est un groupe donneur d'électrons), le produit est donc préférentiellement alkylé. Si l'on veut obtenir des produits de monoalkylation, il faut alors prélever un large excès du composé aromatique.
2) Comme la sulfonation, la réaction d'alkylation de Friedel-Crafts réversible(voir également la section IV.1.D).
3) Même dans des conditions douces, les halogénures d'alkyle primaires et secondaires donnent respectivement préférentiellement des alkylarènes secondaires ou tertiaires, car l'alkylation se produit dans des conditions proches de SN 1 réactions (note 37) Le regroupement peut être évité si vous travaillez à basse température.
L'acylation Friedel-Crafts des composés aromatiques est la méthode la plus importante pour la synthèse des cétones aromatiques grasses. Les dérivés de l'acide carboxylique, tels que les halogénures et anhydrides d'acyle, possèdent un groupe carbonyle polaire et sont en principe capables de substitution électrophile dans les systèmes aromatiques :

L'activité électrophile de ces composés est cependant faible et doit être renforcée par l'action des acides de Lewis. Dans ce cas, le catalyseur acide, en règle générale, attaque l'atome d'oxygène composé carbonylé et, en déplaçant la densité électronique, augmente la charge positive de l'atome de carbone voisin. En conséquence, il se forme un complexe polarisé (et, à la limite, un cation acyle), agissant comme un électrophile :


Une différence importante entre la réaction d'acylation avec des halogénures d'acyle et la réaction d'alkylation avec des halogénures d'alkyle est que dans la première de ces réactions plus de 1 mol d'acide de Lewis est nécessaire, tandis que dans le second, seules des quantités catalytiques sont nécessaires. Cela est dû au fait que l'acide de Lewis forme un complexe à la fois avec le dérivé acylant de l'acide carboxylique et avec la cétone, le produit de la réaction. Lors de la réaction avec les anhydrides, l'acide résultant se lie à une autre mole de catalyseur, de sorte qu'au total, au moins deux moles sont nécessaires. Dans chaque cas, à la fin de la réaction, le complexe cétonique résultant avec le chlorure d'aluminium (ou un autre acide de Lewis) doit être détruit par hydrolyse (acide chlorhydrique avec de la glace).
La polyacylation n'est pas observée car la cétone résultante est significativement moins réactive que le composé parent (voir section IV.1.B). Par conséquent, il est souvent préféré d'obtenir des alkylbenzènes non pas par alkylation directe, mais par acylation de Friedel-Crafts suivie d'une réduction. Les composés aromatiques avec des substituants fortement désactivants, tels que les groupes nitro ou cyano, ne sont pas non plus acylés par Friedel-Crafts.
Tâches de contrôle
2. Dessinez un diagramme d'énergie potentielle pour une réaction de substitution aromatique électrophile dans laquelle l'étape lente est la formation
-complexe (par exemple, nitration du benzène avec du borofluorure de nitronium ;
voir section IV.1.A).
3. Quel produit se forme principalement lors de la bromation : a) paire-nitrotoluène; b) méta-les acides nitrobenzènesulfoniques ; V) ortho-nitrophénol.
4. L'adrénaline (1-(3,4"-dihydroxyphényl)-2-méthylaminoéthanol) est la première hormone isolée de la médullosurrénale ; elle est actuellement synthétisée en trois étapes à partir du pyrocatéchol. Écrivez l'équation de la première étape de cette synthèse - la réaction d'acylation du pyrocatéchol (1,2-dihydroxybenzène) avec le chlorure d'acide chloroacétique et expliquez le mécanisme).
5. L'une des réactions qualitatives aux protéines est la réaction xanthoprotéique, indiquant la présence d'acides aminés aromatiques. Il s’agit de traiter la protéine avec de l’acide nitrique lorsqu’elle est chauffée. Écrivez l’équation de la réaction de la xanthoprotéine avec la tyrosine (voir section I), formée à la suite de l’hydrolyse des protéines.
La nitration du benzène peut être réalisée avec de petites quantités de matières premières sans isoler un produit pur. Pour obtenir du nitrobenzène en utilisant l'équation :
C 6 H 6 + HNO 3 à C 6 H 5 NO 2+ H2O
de l'acide nitrique concentré (densité spécifique 1,4) est nécessaire. Le mélange réactionnel ne doit pas être chauffé au-dessus de 50-60°C. Lorsque de l'acide dilué est utilisé, la réaction de nitration ne se produit pas ; avec l'augmentation de la température, une formation notable de dinitrobenzène commence.
Il résulte de l’équation que la réaction nécessite des quantités équimoléculaires des matières premières. Cependant, dans ce cas, la réaction ne sera pas complète, car l’eau libérée diluera l’acide nitrique et perdra sa propriété nitrant. Par conséquent, pour achever la réaction, il est nécessaire de prendre plus d’acide nitrique que ce qui devrait l’être selon la théorie. Mais pour éviter que la réaction ne devienne trop violente, il faut dissoudre l'acide nitrique dans de l'acide sulfurique concentré, ce qui ne prive pas l'acide nitrique de son effet nitrant et fixe l'eau libérée lors de la réaction.
Pour éviter toute possibilité d'augmentation de la température pendant la réaction, ne mélangez pas toutes les substances en même temps, mais ajoutez progressivement du benzène au mélange d'acides. 8 ml d'acide sulfurique concentré et 5 ml d'acide nitrique concentré sont versés dans un petit flacon. Refroidissez le mélange sous l'eau courante. Ensuite, 4 ml de benzène sont ajoutés au mélange refroidi par petites portions, en agitant constamment le ballon pour obtenir un meilleur mélange de liquides qui ne se dissolvent pas les uns dans les autres (le mélange d'acides constitue la couche inférieure, le benzène constitue la couche supérieure). . Après avoir ajouté tout le benzène pour que la réaction soit complète, le ballon est fermé par un bouchon à tube vertical (la vapeur de benzène est volatile) et chauffé dans un bain-marie préchauffé à 60°C.
Secouez le flacon de temps en temps pour mieux mélanger les liquides.
La durée du chauffage peut être déterminée non pas tant par la nécessité d'atteindre l'intégralité de la réaction, mais par le temps disponible pendant la leçon. Lorsque vous travaillez dans une tasse, le chauffage doit continuer pendant 30 à 40 minutes. Dans la leçon, il est possible de démontrer la formation de nitrobenzène après 10 minutes de chauffage et même sans chauffage supplémentaire, si la réaction s'est bien déroulée lorsque le benzène a été ajouté à un mélange d'acides.
Le nitrobenzène est placé en couche sur le mélange acide. Versez le contenu du flacon dans un verre rempli d'eau. Dans ce cas, les acides se dissolvent dans l’eau, tandis que le nitrobenzène s’accumule au fond du verre sous la forme d’un liquide lourd jaunâtre. Si le temps le permet, égouttez une partie du liquide du nitrobenzène et séparez-le à l'aide d'une ampoule à décanter.
Lorsque des quantités importantes de nitrobenzène sont obtenues et qu'il est nécessaire de le purifier, le nitrobenzène est lavé avec de l'eau, une solution alcaline diluée (5 %), puis à nouveau avec de l'eau, en séparant à chaque fois les liquides à l'aide d'une ampoule à décanter. Le nitrobenzène est ensuite déshydraté en le chauffant avec du chlorure de calcium granulaire jusqu'à ce que le liquide devienne clair. Un chauffage est nécessaire pour réduire la viscosité du nitrobenzène et ainsi obtenir un contact plus complet avec le chlorure de calcium. Enfin, le nitrobenzène peut être distillé à partir d'un petit flacon équipé d'un refroidisseur d'air à une température de 204 à 207°C. Pour éviter la décomposition des résidus de dinitrobenzène, la distillation à sec n'est pas recommandée.
Les propriétés physiques du méthylbenzène (toluène) sont similaires à celles du benzène. Dans des conditions normales, c'est un liquide incolore, insoluble dans l'eau, mais soluble dans les solvants organiques. Comme le benzène, c'est un bon solvant pour les composés organiques. Actuellement, le toluène est plus largement utilisé comme solvant que le benzène en raison de sa toxicité beaucoup plus faible.
Propriétés chimiques
Toutes les réactions du méthylbenzène peuvent être divisées en deux types : a) les réactions impliquant le cycle benzénique et b) les réactions impliquant le groupe méthyle.
Réactions dans le cycle aromatique. Le méthylbenzène subit toutes les réactions de substitution électrophile décrites ci-dessus pour le benzène, à savoir : la nitration, l'halogénation, la sulfonation et la réaction de Friedel-Crafts. Dans toutes ces réactions, le méthylbenzène présente une réactivité plus élevée et ses réactions se déroulent plus rapidement.
La nitration du méthylbenzène peut être réalisée de la même manière que celle du benzène. Le produit de nitration du méthylbenzène est un mélange de deux isomères du méthylnitrobenzène :

La chloration du toluène (méthylbenzène) dans le cycle benzénique peut être réalisée en faisant passer du chlore gazeux à travers le toluène en présence de chlorure d'aluminium (la réaction est effectuée dans l'obscurité). Le chlorure d'aluminium joue dans ce cas le rôle de catalyseur. Dans ce cas, des isomères 2 et 4-substitués se forment :

La sulfonation du méthylbenzène avec de l'acide sulfurique concentré conduit également à la formation d'un mélange d'isomères 2 et 4-substitués :

Le mécanisme de toutes ces réactions de substitution électrophile est similaire au mécanisme des réactions correspondantes du benzène. Dans ces réactions, des isomères 3-substitués se forment en si petites quantités qu’ils peuvent être négligés. Ci-dessous, cette fonctionnalité sera discutée en détail en considérant la capacité de direction (d'orientation) des groupes de substitution.
Réactions en chaîne secondaire. Le groupe méthyle du méthylbenzène peut subir certaines réactions caractéristiques des alcanes, mais aussi d'autres réactions non caractéristiques des alcanes. Examinons un exemple de chacune de ces réactions.
Comme les alcanes, le groupe méthyle peut être halogéné par un mécanisme radicalaire. Pour effectuer cette réaction, le chlore est soufflé à travers du méthylbenzène bouillant en présence de la lumière du soleil ou d’une source de lumière ultraviolette. A noter que l'halogénation du cycle benzénique dans le méthylbenzène nécessite des conditions complètement différentes.

Veuillez noter que cette réaction est une substitution. Une halogénation supplémentaire conduit à la formation des composés suivants :

La bromation du méthylbenzène s'effectue dans des conditions similaires et conduit à la formation des composés de substitution du brome correspondants.
La section précédente indiquait que les alcanes n'entrent pas dans des réactions d'oxydation même avec des agents oxydants aussi puissants que le permanganate de potassium. Cependant, la chaîne latérale méthyle du méthylbenzène est sujette à l'oxydation même par des agents oxydants relativement doux comme l'oxyde

Des agents oxydants plus puissants, tels que le permanganate de potassium, provoquent une oxydation supplémentaire :

Effet directeur (orientant) des substituants sur le cycle benzénique
Nous avons déjà indiqué plus haut que la substitution électrophile du méthylbenzène conduit à la formation d'isomères 2- et 4-substitués du méthylbenzène. Par conséquent, le groupe méthyle du méthylbenzène est appelé groupe directeur 2,4 (ou, autrement, orienté vers les positions 2 et 4). Il existe d'autres substituants sur le cycle benzénique qui ont également un effet directeur 2,4 dans les réactions de substitution électrophile. Pour de telles réactions, l’équation générale suivante peut être écrite :

Dans cette équation signifie électrophile et X-2,4 est un substituant directeur. Ces substituants sont généralement des groupes saturés. Ceux-ci incluent. Dans certaines conditions, les substituants dirigeant 2,4 s'avèrent également être dirigeant 6 :

Les réactions de substitution électrophile aux positions 2 et 4 ont généralement une vitesse plus élevée que les réactions correspondantes dans le benzène.

Ceux-ci incluent des groupes tels que et. Les réactions de substitution électrophile en position 3 se déroulent plus lentement que les réactions correspondantes du benzène.
La capacité de direction (d'orientation) des substituants dépend du fait qu'ils soient donneurs d'électrons pour le cycle benzénique ou, à l'inverse,
en éloigner les électrons. La substitution 2,4 est provoquée par un effet inductif positif ou un effet mésomère positif. -Les groupes directeurs donnent des électrons au cycle benzénique, provoquant ces effets, et activent ainsi le cycle. C’est pourquoi on les appelle des groupes activateurs. 3-Les groupes directeurs éloignent les électrons du cycle benzénique, provoquant - et - des effets. C’est ce qu’on appelle des groupes de désactivation.
Alors redisons-le !
1. Les composés aromatiques ont les propriétés générales suivantes : a) brûlent avec formation d'une flamme fumée ;
(voir scan)
Riz. 18.7. Les réactions les plus importantes du benzène.

Riz. 18.8. Les réactions les plus importantes du méthylbenzène (toluène).
b) subir des réactions de substitution,
c) subir difficilement des réactions d'addition.
2. La molécule de benzène peut être considérée comme un hybride résonant formé de deux structures résonantes limitantes.
3. Les réactions chimiques les plus importantes du benzène sont présentées sur la Fig. 18.7.
4. Les réactions chimiques les plus importantes du méthylbenzène sont illustrées à la Fig. 18.8.
5. Dans les réactions de condensation, deux molécules en réaction se combinent en une nouvelle molécule avec l'élimination simultanée d'une petite molécule d'un composé simple, comme l'eau ou le chlorure d'hydrogène.
6. Les substituants saturés sur le cycle benzénique ont un effet directeur (orientant) 2,4.
7. Les substituants insaturés sur le cycle benzénique ont un effet directeur (orientant).

L'existence de complexes - en tant qu'intermédiaires stables a été prouvée par des expériences directes. Cela n'indique pas nécessairement que l'état de transition de la réaction d'addition-élimination électrophile est de structure similaire à celle du complexe -. En effet, le rôle des complexes dans ces réactions fait encore débat. Cette section se concentrera sur le mécanisme de nitration aromatique, qui a suscité beaucoup d’intérêt. D'autres réactions électrophiles d'addition-élimination listées ci-dessus seront alors envisagées.
Les résultats de tous les travaux cinétiques, spectroscopiques et cryoscopiques publiés avant 1960 confortent l'idée selon laquelle l'ion nitronium est un électrophile efficace dans les réactions de nitration des arènes. À l’aide de méthodes spectroscopiques et cryoscopiques, de faibles concentrations d’ions nitronium ont été détectées dans l’acide nitrique anhydre. Il est également établi de manière fiable qu'en présence d'un large excès d'acide sulfurique, l'acide nitrique est complètement transformé en bisulfate de nitronium. Cependant, la nitration des composés aromatiques, dont la réactivité est similaire à celle du benzène, se produit en présence de quantités d'eau telles que les ions nitronium ne peuvent pas être détectés. Parallèlement, la participation des ions nitronium à la nitration est également strictement démontrée en comparant le taux de nitration avec le taux d'échange 180 entre le milieu et l'acide nitrique. Les réactions de nitration de certains substrats réactifs en présence d'eau ont un ordre cinétique nul. Cela indique que l'agent nitrant est formé à partir de l'acide nitrique dans une étape lente (limitante), précédant l'attaque de l'électrophile par le cycle aromatique. En l'absence de substrat aromatique, le taux d'échange 180 entre l'eau et l'acide nitrique est d'ordre zéro, comme pour la nitration. Ces résultats sont mieux expliqués par le diagramme suivant (équation 28) :
L'isolement de sels de nitronium contenant des anions perchlorate, tétrafluoroborate et hexafluorophosphate, et le fait que ces sels sont des agents nitrants efficaces, confortent également les conclusions tirées précédemment.
La nitration a été utilisée pour établir la réactivité relative d'un grand nombre de composés aromatiques. Les taux relatifs de substitution aux positions et et - avec corrections statistiques, appelés facteurs de taux partiels, ont été calculés pour un grand nombre de benzènes monosubstitués. Les calculs ont été effectués sur la base de taux relatifs en utilisant des données cinétiques ou par la méthode des réactions compétitives en déterminant la composition isomère du produit de réaction dans les mêmes conditions expérimentales. Des résultats très similaires ont été obtenus par nitration de mélanges équimolaires de benzène et de toluène avec de l'acide nitrique dans du nitrométhane, de l'acétonitrile, de l'anhydride acétique ou des solvants acides. Ces résultats confirment également le caractère universel du mécanisme impliquant le cation nitronium. Dans le tableau 2.5.2 montre des exemples typiques. Il est évident que bien que la distribution des isomères dans les réactions avec des sels de nitronium pré-préparés soit proche des résultats obtenus précédemment, les taux relatifs dans les réactions avec les sels sont beaucoup plus proches les uns des autres et, par conséquent, le facteur de taux partiel, par exemple pour le méta -l'attaque en position, est apparemment plus faible que pour les positions individuelles du benzène.
Tableau 2.5.2. Facteurs d'orientation, de réactivité relative et de taux partiel dans la nitration de certaines molécules typiques

Vous trouverez ci-dessous une méthode de calcul utilisant l'exemple de la nitration du toluène par rapport au benzène dans l'acide acétique à . Rappelons que le benzène a six positions équivalentes et que le toluène a une position para et deux positions ortho et méta :

L'étude de la nitration de mélanges de benzène avec du toluène à l'aide de sels de nitronium pré-préparés (voir tableau 2.5.2) et des expériences similaires avec le m-xylène et le mésitylène, dont la réactivité relative par rapport au benzène s'est avérée être de 1,7 et 2,7, respectivement, augmenté le nombre de questions peu claires. Il a déjà été mentionné ci-dessus les résultats incompréhensibles obtenus lors du calcul du facteur de vitesse partiel pour la méta-position. De plus, il a été noté que dans le cas où la réaction a le temps de se produire principalement avant que les réactifs ne forment un mélange suffisamment homogène, les résultats de la détermination de la réactivité à partir d'expériences de déplacement compétitif peuvent être erronés.
Évidemment, cette réactivité anormale n'est pas confirmée lors de la détermination des constantes de vitesse de nitration avec des sels de nitronium à l'aide de méthodes cinétiques. Cependant, des tentatives ont été faites pour assurer un mélange suffisant des réactifs en effectuant la réaction à différentes concentrations. Ces résultats ont conduit à l'hypothèse que la vitesse d'un certain nombre de réactions des arènes avec des électrophiles, y compris la vitesse de nitration avec les sels de nitronium, est liée à la -basicité plutôt qu'à la -basicité des arènes. En d’autres termes, la formation du complexe β est l’étape limitante. Cependant, la localisation de l'électrophile est contrôlée par la -basicité, comme le montrent les données disponibles sur la distribution des isomères. Cette vision de la nitration aromatique a de nouveau été explorée en détail. Il a été noté que la corrélation entre le rapport des produits de nitration avec les sels de nitronium et la stabilité relative des méthylbenzènes protonés n'est apparemment pas pire que la corrélation avec la stabilité des complexes -. Le rapport des produits de nitration compétitive des polyméthylbenzènes avec un mélange d'acides dans le sulfolane est également mal corrélé à la stabilité des -complexes.
La question liée au degré de réaction lors du mélange des réactifs a été étudiée à l'aide de l'exemple de la nitration du bibenzyle, dans laquelle chaque cycle a une réactivité similaire à celle du toluène, et, en outre, du transfert de l'influence du substituant entre les cycles. est minime. Lorsque des concentrations équimolaires de sel de bibenzyle et de nitronium sont utilisées et que les réactifs sont complètement mélangés avant le début de la réaction, du mononitrobibenzyle (50 %) et du dinitrobibenzyle doivent se former. D'un autre côté, si les réactifs ne sont pas complètement mélangés avant la réaction, la quantité de substrat n'ayant pas réagi et de dinitrobibenzyle devrait augmenter et la quantité de mononitrobibenzyle devrait diminuer. Cependant, cette idée est trop simplifiée, ce qui découle clairement des résultats de la nitration du bibenzyle avec de l'acide nitrique dans l'anhydride acétique. L'intégralité du mélange n'est pas importante pour ce système, mais la quantité de dinitrobeisil ne représente que 55 % de celle attendue. Lors de la nitration du bibenzyle dans le sulfolane avec du tétrafluoroborate de nitronium en utilisant diverses concentrations et conditions de mélange, la quantité de produit disubstitué était toujours significativement supérieure à celle obtenue à partir du calcul ci-dessus. Ainsi, la méthode simple des réactions compétitives n'est pas adaptée pour déterminer la réactivité relative des composés aromatiques lors de la nitration avec des sels de nitronium. Évidemment, la vitesse de mélange est la principale influence.
Des résultats intéressants ont également été obtenus en étudiant la cinétique de nitration des arènes dont la réactivité est supérieure à celle du benzène et du toluène.
On pourrait s'attendre à ce que la réactivité du -xylène, du -xylène et du mésitylène soit nettement supérieure à celle du benzène (facteurs autour de et ), cependant, si l'on considère les résultats de la nitration dans l'acide sulfurique aqueux (68,3%), il s'avère que ce facteur est limité à une valeur de 40. On suppose que l'étape limitante dans ces réactions est la formation d'une paire collisionnelle entre un ion nitronium et un substrat aromatique. Quelle est la différence entre cette hypothèse et l’hypothèse d’un complexe de formation limitant le taux ? Il a été noté précédemment que le rapport entre les produits n’est pas bien corrélé à la stabilité des complexes β. Ces faits peuvent être expliqués sans recourir à des interactions attractives au sein d’une « paire de collision ». La barrière énergétique pour la dissociation d'une « paire de collision » en composants peut être plus élevée dans un environnement acide ; cette énergie est tout à fait suffisante pour la formation sélective d'un complexe. Puisque les données cinétiques sont cohérentes avec la théorie de diffusion des collisions, le terme « paire de collisions » doit être préféré. Une discussion de toutes les données ci-dessus conduit à la conclusion que le schéma d'équations (28) doit être modifié comme le montre l'équation (29), qui ne détermine la vitesse que dans des circonstances exceptionnelles. Les différentes constantes de vitesse d'attaque aux positions ortho, méta ou para indiquent que la distribution des isomères ne dépend pas du stade limite, puisque la « paire de collision » comprend la molécule d'arène entière, et non ses atomes individuels.

La discussion sur la nitration peut être facilement résumée en termes de profils énergétiques, comme le montre la Fig. 2.5.1. Les résultats expérimentaux montrent que dans le cas de la nitration du benzène et du toluène, l'étape déterminante du taux est la formation du complexe (Fig. a et b, respectivement), et la formation d'une « paire de collisions » détermine le taux de nitration. de pseudocumène (-triméthylbenzène) (Fig. c).
La répartition des isomères découverte lors de la nitration du toluène (voir tableau 2.5.2.) est fondamentalement indépendante des conditions de réaction, à l'exclusion de l'influence habituelle de la température. Cependant, les résultats inhabituels obtenus lors de l'étude de certains polyméthylbenzènes compliquent la compréhension de la réaction de nitration. Ainsi, la nitration avec de l'acide nitrique dans l'anhydride acétique conduit à des produits d'acétoxylation. Cela mélange les choses, suggérant qu’une acétoxylation électrophile a lieu. Ce n’est actuellement pas le cas.


Riz. 2.5.1. Profils énergétiques pour les réactions de nitration du benzène (a) et du toluène (b) et la réaction de formation de 5- et : a, b-l'étape limite est la formation du -complexe ; c - l'étape limite est la formation d'un couple de collision.
Bien que l’acide nitrique soit présent principalement sous forme de nitrate d’acétyle, des doutes subsistent quant à la véritable nature de cet électrophile. Ainsi, l'acide nitrique dans l'anhydride acétique réagit avec le -xylène pour former un mélange de -diméthylnitrobenzène et de -diméthylphénylacétate. L'isolement des adduits dans lesquels le nitrate d'acétyle est ajouté en position de substitution (attaque ipso), et le fait que ces adduits se décomposent apparemment par un mécanisme intramoléculaire pour former de l'acétate (57), plaident en faveur du mécanisme présenté dans le diagramme d'équation. (30) :

Des adduits similaires (58)-(64) ont été isolés dans des réactions avec d'autres arènes, et certains adduits ont été trouvés comme intermédiaires. En faisant réagir de l'acide nitrique dans de l'anhydride acétique avec du n-tert-butyltoluène à basse température, trois produits d'addition ont été obtenus et isolés. Le produit principal est l'adduit α (64), qui semble être formé en plus grande quantité que les deux autres isomères en raison d'un obstacle stérique en position 4 (ipso à tert-Vi). D'autres nucléophiles, tels que l'ion nitrate et l'eau, peuvent également s'ajouter au complexe α ipso-substitué. Les complexes β ipso-substitués peuvent être régénérés par acidolyse.

La nitration de l'o-xylène dans l'acide sulfurique donne du -diméthylnitrobenzène et du -diméthylnitrobenzène dont le rendement dépend de l'acidité du système. À faible acidité, des nitrodiméthylphénols sont également obtenus. L'acidolyse des esters (58) et (59) produit du -diméthylnitrobenzène dont le rendement augmente avec l'acidité ; cela conforte la suggestion selon laquelle la nitration de la position ortho du groupe alkyle implique probablement souvent une attaque ipso initiale. Le fait que l’acidolyse (58) et (59) ne produisent pas de -diméthylnitrobenzène signifie que le complexe o original ipso-substitué ne se transforme pas à nouveau en une « paire de collision » et que ni les déplacements α ni les déplacements β successifs du groupe nitro ne se produisent. dedans. Ces résultats sont résumés dans les équations (31). L'augmentation du rendement et l'augmentation de l'acidité du milieu lors de la nitration du -triméthylbenzène dans l'acide sulfurique sont attribuées à la migration du groupe nitro de vers et de vers, respectivement.

Actuellement, aucun exemple fiable de déplacements intramoléculaires séquentiels via une position non substituée n’est connu. Cependant, des déplacements successifs d'une position ipso à une autre position ipso sont connus. Par exemple, l'acidolyse de (65) produit des isomères structuraux (66) et (67), comme le montre l'équation (32).

Auparavant, l'élimination du substituant ipso était évoquée, par exemple dans le cas de la protodésilylation et de la protodésulfuration (voir pp. 330, 331). De ce point de vue, nous discuterons uniquement du remplacement du substituant ipso par un groupe nitro. De nombreuses réactions de ce type sont connues depuis longtemps, mais leur mécanisme n'est généralement pas clair. De telles réactions comprennent la désalkylation, la désacylation, la désilylation, la désulfonation, la décarboxylation, la dédiazotation et la déshalogénation. Les réactions impliquant des substrats hautement activés vers une attaque électrophile peuvent impliquer une nitrosation initiale suivie d'une oxydation.