Systèmes électrochimiques
caractéristiques générales
Électrochimie - une branche de la chimie qui étudie les processus d'apparition de différences de potentiel et de conversion de l'énergie chimique en énergie électrique (cellules galvaniques), ainsi que la mise en œuvre de réactions chimiques dues à la dépense d'énergie électrique (électrolyse). Ces deux procédés, de nature commune, sont largement utilisés dans la technologie moderne.
Les cellules galvaniques sont utilisées comme sources d'énergie autonomes et de petite taille pour les machines, les appareils radio et les appareils de commande. Grâce à l'électrolyse, diverses substances sont obtenues, les surfaces sont traitées et des produits de la forme souhaitée sont créés.
Les processus électrochimiques ne profitent pas toujours aux humains et causent parfois de graves dommages, provoquant une corrosion accrue et la destruction des structures métalliques. Afin d’utiliser habilement les procédés électrochimiques et de lutter contre les phénomènes indésirables, il faut les étudier et pouvoir les réguler.
La cause des phénomènes électrochimiques est le transfert d'électrons ou un changement dans l'état d'oxydation des atomes de substances participant aux processus électrochimiques, c'est-à-dire des réactions redox se produisant dans des systèmes hétérogènes. Dans les réactions redox, les électrons sont directement transférés de l’agent réducteur à l’agent oxydant. Si les processus d'oxydation et de réduction sont spatialement séparés et que les électrons sont dirigés le long d'un conducteur métallique, alors un tel système représentera une cellule galvanique. La raison de l'apparition et du flux du courant électrique dans une cellule galvanique est la différence de potentiel.
Le potentiel de l'électrode. Potentiels des électrodes de mesure
Si vous prenez une plaque de n'importe quel métal et que vous la plongez dans l'eau, les ions de la couche superficielle, sous l'influence des molécules d'eau polaires, se détachent et s'hydratent dans le liquide. À la suite de cette transition, le liquide se charge positivement et le métal négativement, puisqu'un excès d'électrons y apparaît. L’accumulation d’ions métalliques dans le liquide commence à inhiber la dissolution du métal. Un équilibre mobile s’établit
Me 0 + mH 2 O = Me n + × m H 2 O + ne -
L'état d'équilibre dépend à la fois de l'activité du métal et de la concentration de ses ions en solution. Dans le cas des métaux actifs dans la série de tension jusqu'à l'hydrogène, l'interaction avec les molécules d'eau polaires se termine par la séparation des ions métalliques positifs de la surface et la transition des ions hydratés en solution (Fig. b). Le métal devient chargé négativement. Le processus est l’oxydation. À mesure que la concentration d’ions près de la surface augmente, le processus inverse devient possible : la réduction des ions. L’attraction électrostatique entre les cations en solution et les électrons en excès à la surface forme une double couche électrique. Cela conduit à l'apparition d'une certaine différence de potentiel, ou saut de potentiel, à l'interface entre le métal et le liquide. La différence de potentiel qui apparaît entre un métal et son environnement aqueux est appelée le potentiel de l'électrode. Lorsqu’un métal est immergé dans une solution d’un sel de ce métal, l’équilibre se déplace. L'augmentation de la concentration d'ions d'un métal donné dans la solution facilite le processus de transition des ions de la solution au métal. Les métaux dont les ions ont une capacité importante à passer en solution seront chargés positivement dans une telle solution, mais dans une moindre mesure que dans l'eau pure.
Pour les métaux inactifs, la concentration d’équilibre des ions métalliques en solution est très faible. Si un tel métal est immergé dans une solution d'un sel de ce métal, des ions chargés positivement sont libérés sur le métal à un rythme plus rapide que la transition des ions du métal à la solution. La surface métallique recevra une charge positive et la solution recevra une charge négative en raison de l'excès d'anions de sel. Et dans ce cas, une double couche électrique apparaît à l’interface métal-solution, d’où une certaine différence de potentiel (Fig. c). Dans le cas considéré, le potentiel de l'électrode est positif.
Riz. Le processus de transition d'un ion d'un métal à une solution :
un équilibre; b- dissolution ; c – dépôt
Le potentiel de chaque électrode dépend de la nature du métal, de la concentration de ses ions dans la solution et de la température. Si un métal est immergé dans une solution de son sel contenant une mole d'ion métallique pour 1 dm 3 (dont l'activité est de 1), alors le potentiel de l'électrode sera une valeur constante à une température de 25 o C et une pression de 1 au m. Ce potentiel est appelé potentiel d'électrode standard (Eo).
Les ions métalliques ayant une charge positive, pénétrant dans la solution et se déplaçant dans le champ potentiel de l'interface métal-solution, dépensent de l'énergie. Cette énergie est compensée par le travail de dilatation isotherme d’une concentration d’ions plus élevée en surface vers une concentration plus faible dans la solution. Les ions positifs s'accumulent dans la couche superficielle jusqu'à une concentration Avec Ô, puis passez en solution, où la concentration d'ions libres Avec. Le travail du champ électrique EnF est égal au travail de dilatation isotherme RTln(с o /с). En assimilant les deux expressions du travail, nous pouvons déduire l’ampleur du potentiel
En F = RTln(s o /s), -E = RTln(s/s o)/nF,
où E est le potentiel métallique, V ; R – constante universelle des gaz, J/mol K ; T – température, K ; n – charge ionique ; F – Numéro Faraday ; с – concentration d'ions libres ;
с о – concentration d'ions dans la couche superficielle.
Il n'est pas possible de mesurer directement la valeur du potentiel, car il est impossible de déterminer expérimentalement la valeur du potentiel. Les valeurs des potentiels d'électrode sont déterminées empiriquement par rapport à la valeur d'une autre électrode dont le potentiel est classiquement supposé nul. Une telle électrode étalon ou de référence est électrode à hydrogène normale (n.v.e.) . La structure de l'électrode à hydrogène est représentée sur la figure. Il se compose d'une plaque de platine recouverte de platine déposée électrolytiquement. L'électrode est immergée dans une solution 1M d'acide sulfurique (l'activité des ions hydrogène est de 1 mol/dm3) et est lavée par un courant d'hydrogène gazeux sous une pression de 101 kPa et T = 298 K. Lorsque le platine est saturé d'hydrogène , l'équilibre s'établit sur la surface métallique, le processus global est exprimé par l'équation
2Н + +2е ↔ Н 2 .
Si une plaque de métal immergée dans une solution 1M d'un sel de ce métal est reliée par un conducteur externe à une électrode à hydrogène standard, et que les solutions sont reliées par une clé électrolytique, alors on obtient une cellule galvanique (Fig. 32). La force électromotrice de cette cellule galvanique sera la quantité potentiel d'électrode standard d'un métal donné (E Ô ).

Schéma de mesure du potentiel d'électrode standard
par rapport à l'électrode à hydrogène
En prenant du zinc dans une solution 1 M de sulfate de zinc comme électrode et en le connectant avec une électrode à hydrogène, on obtient une cellule galvanique dont le circuit s'écrira comme suit :
(-) Zn/Zn 2+ // 2H + /H 2, Pt (+).
Dans le diagramme, une ligne indique l'interface entre l'électrode et la solution, deux lignes indiquent l'interface entre les solutions. L'anode est inscrite à gauche, la cathode à droite. Dans un tel élément, la réaction Zn o + 2H + = Zn 2+ + H 2 a lieu et les électrons traversent le circuit externe du zinc à l'électrode à hydrogène. Potentiel d'électrode standard pour électrode de zinc (-0,76 V).
En prenant une plaque de cuivre comme électrode, dans les conditions spécifiées en combinaison avec une électrode à hydrogène standard, on obtient une cellule galvanique
(-) Pt, H 2 /2H + //Cu 2+ /Cu (+).
Dans ce cas, la réaction se produit : Cu 2+ + H 2 = Cu o + 2H +. Les électrons se déplacent à travers le circuit externe de l'électrode à hydrogène vers l'électrode à cuivre. Potentiel d'électrode standard de l'électrode de cuivre (+0,34 V).
Un certain nombre de potentiels d'électrodes standard (tensions). équation de Nernst
En disposant les métaux par ordre croissant de leurs potentiels d'électrode standard, on obtient une série de tensions de Nikolai Nikolaevich Beketov (1827-1911), ou une série de potentiels d'électrode standard. Les valeurs numériques des potentiels d'électrodes standard pour un certain nombre de métaux techniquement importants sont données dans le tableau.
Plage de contrainte du métal

Un certain nombre de contraintes caractérisent certaines propriétés des métaux :
1. Plus le potentiel d'électrode d'un métal est bas, plus il est chimiquement actif, plus il est facile à oxyder et plus il est difficile de récupérer ses ions. Les métaux actifs dans la nature n'existent que sous forme de composés Na, K, ..., se retrouvent dans la nature aussi bien sous forme de composés qu'à l'état libre de Cu, Ag, Hg ; Au, Pt - uniquement à l'état libre ;
2. Les métaux qui ont un potentiel d’électrode plus négatif que le magnésium déplacent l’hydrogène de l’eau ;
3. Les métaux qui se trouvent dans la série de tension avant l'hydrogène déplacent l'hydrogène des solutions d'acides dilués (dont les anions ne présentent pas de propriétés oxydantes) ;
4. Chaque métal de la série qui ne décompose pas l'eau déplace les métaux qui ont des valeurs de potentiels d'électrode plus positives des solutions de leurs sels ;
5. Plus les métaux diffèrent dans les valeurs des potentiels d'électrode, plus la valeur emf est élevée. une cellule galvanique sera construite à partir d'eux.
La dépendance du potentiel d'électrode (E) sur la nature du métal, l'activité de ses ions en solution et la température est exprimée par l'équation de Nernst
E Me = E o Me + RTln(a Me n +)/nF,
où E o Me est le potentiel d'électrode standard du métal et Men + est l'activité des ions métalliques en solution. A une température standard de 25 o C, pour les solutions diluées, en remplaçant l'activité (a) par la concentration (c), le logarithme népérien par un décimal et en substituant les valeurs de R, T et F, on obtient
E Me = E o Me + (0,059/n)logс.
Par exemple, pour une électrode de zinc placée dans une solution de son sel, la concentration en ions hydratés Zn 2+ × mH 2 O Abrégeons-le en Zn 2+ , alors
E Zn = E o Zn + (0,059/n) log[ Zn 2+ ].
Si = 1 mol/dm 3, alors E Zn = E o Zn.
Cellules galvaniques, leur force électromotrice
Deux métaux immergés dans des solutions de leurs sels, reliés par un conducteur, forment une cellule galvanique. La première cellule galvanique a été inventée par Alexandre Volt en 1800. La cellule était constituée de plaques de cuivre et de zinc séparées par un tissu imbibé d'une solution d'acide sulfurique. Lorsqu'un grand nombre de plaques sont connectées en série, l'élément Volta a une force électromotrice (fem) importante.
L'apparition d'un courant électrique dans une cellule galvanique est provoquée par la différence des potentiels d'électrode des métaux prélevés et s'accompagne de transformations chimiques se produisant au niveau des électrodes. Considérons le fonctionnement d'une cellule galvanique en utilisant l'exemple d'une cellule cuivre-zinc (J. Daniel - B. S. Jacobi).

Schéma d'une cellule galvanique cuivre-zinc Daniel-Jacobi
Sur une électrode de zinc immergée dans une solution de sulfate de zinc (c = 1 mol/dm 3), l'oxydation du zinc (dissolution du zinc) se produit Zn o - 2e = Zn 2+. Les électrons entrent dans le circuit externe. Le Zn est une source d'électrons. La source des électrons est considérée comme l’électrode négative – l’anode. Sur une électrode de cuivre immergée dans une solution de sulfate de cuivre (c = 1 mol/dm 3), les ions métalliques sont réduits. Des atomes de cuivre se déposent sur l'électrode Cu 2+ + 2e = Cu o. L'électrode de cuivre est positive. C'est la cathode. Dans le même temps, certains ions SO 4 2- traversent le pont salin dans un récipient contenant une solution de ZnSO 4 . En additionnant les équations des processus se produisant à l'anode et à la cathode, nous obtenons l'équation totale
|
Boris Semenovitch Jacobi (Moritz Hermann) (1801-1874) |

ou sous forme moléculaire
Il s’agit d’une réaction redox courante se produisant à l’interface métal-solution. L'énergie électrique d'une cellule galvanique est obtenue par une réaction chimique. La cellule galvanique considérée peut s'écrire sous la forme d'un bref circuit électrochimique
(-) Zn/Zn 2+ //Cu 2+ /Cu (+).
Une condition nécessaire au fonctionnement d'une cellule galvanique est la différence de potentiel, on l'appelle force électromotrice d'une cellule galvanique (emf) . E.m.f. tout élément galvanique en fonctionnement a une valeur positive. Pour calculer la fem. cellule galvanique, il faut soustraire la valeur du potentiel le moins positif de la valeur du potentiel le plus positif. Donc c.e.m. La cellule galvanique cuivre-zinc dans des conditions standard (t = 25 o C, c = 1 mol/dm 3, P = 1 atm) est égale à la différence entre les potentiels d'électrode standard de cuivre (cathode) et de zinc (anode), c'est-à-dire est
f.e.m. = E o C u 2+ / Cu - E o Zn 2+ / Zn = +0,34 V – (-0,76 V) = +1,10 V.
Lorsqu'il est associé au zinc, l'ion Cu 2+ est réduit.
La différence de potentiel d'électrode nécessaire au fonctionnement peut être créée en utilisant la même solution de concentrations différentes et les mêmes électrodes. Une telle cellule galvanique est appelée concentration , et cela fonctionne en égalisant les concentrations de la solution. Un exemple serait une cellule composée de deux électrodes à hydrogène
Pt, H 2 / H 2 SO 4 (s`) // H 2 SO 4 (s``) / H 2, Pt,
où c` = `; c`` = ``.
Si p = 101 kPa, s`< с``, то его э.д.с. при 25 о С определяется уравнением
E = 0,059 lg(s``/s`).
À с` = 1 mol-ion/dm 3 emf. L'élément est déterminé par la concentration d'ions hydrogène dans la deuxième solution, c'est-à-dire E = 0,059 lgс`` = -0,059 pH.
Détermination de la concentration en ions hydrogène et, par conséquent, du pH du milieu par mesure de la force électromotrice. l'élément galvanique correspondant est appelé potentiométrie.
Batteries
Batteries sont appelées cellules galvaniques à action réutilisable et réversible. Ils sont capables de convertir l'énergie chimique accumulée en énergie électrique lors de la décharge et l'énergie électrique en énergie chimique, créant ainsi une réserve pendant la charge. Depuis que l'e.m.f. les batteries sont petites ; pendant le fonctionnement, elles sont généralement connectées aux batteries.
Batterie au plomb . Une batterie au plomb se compose de deux plaques de plomb perforées, dont l'une (négative) après la charge contient une charge - du plomb actif spongieux, et l'autre (positive) - du dioxyde de plomb. Les deux plaques sont immergées dans une solution d'acide sulfurique à 25 - 30 % (Fig. 35). Circuit de batterie
(-) Pb/ p -p H 2 SO 4 / PbO 2 / Pb(+) .
Avant le chargement, une pâte contenant, en plus du liant organique, de l'oxyde de plomb PbO, est enduite dans les pores des électrodes de plomb. À la suite de l'interaction de l'oxyde de plomb avec l'acide sulfurique, du sulfate de plomb se forme dans les pores des plaques d'électrode
PbSO + H 2 SO 4 = PbSO 4 + H 2 O .
Les batteries sont chargées en faisant passer du courant électrique

Processus de décharge

Au total, les processus qui se produisent lors de la charge et de la décharge d'une batterie peuvent être représentés comme suit :
Lors du chargement d'une batterie, la densité de l'électrolyte (acide sulfurique) augmente et lors de la décharge, elle diminue. La densité de l'électrolyte détermine le degré de décharge de la batterie. E.m.f. batterie au plomb 2,1 V.
Avantages batterie au plomb - capacité électrique élevée, fonctionnement stable, un grand nombre de cycles (décharge-charge). Défauts- masse importante et donc faible capacité spécifique, dégagement d'hydrogène lors du chargement, non étanchéité en présence d'une solution d'acide sulfurique concentrée. Les piles alcalines sont meilleures à cet égard.
Piles alcalines. Il s'agit notamment des batteries T. Edison cadmium-nickel et fer-nickel.

Circuits de batterie Edison et de batterie au plomb
|
Thomas Edison(1847-1931) |
Ils se ressemblent. La différence réside dans le matériau des plaques d’électrodes négatives. Dans le premier cas, il s’agit de cadmium, dans le second, de fer. L'électrolyte est une solution de KOH ω = 20% . Les batteries nickel-cadmium sont de la plus grande importance pratique. Schéma de la batterie cadmium-nickel
(-) Solution Cd/KOH/Ni 2 O 3 /Ni (+).
Le fonctionnement d'une batterie cadmium-nickel est basé sur une réaction redox impliquant Ni 3+
E.m.f. d'une batterie nickel-cadmium chargée est de 1,4 V.
Le tableau présente les caractéristiques de la batterie Edison et de la batterie au plomb.
Électrochimie - une branche de la chimie qui étudie les processus d'apparition de différences de potentiel et de conversion de l'énergie chimique en énergie électrique (cellules galvaniques), ainsi que la mise en œuvre de réactions chimiques dues à la dépense d'énergie électrique (électrolyse). Ces deux procédés, de nature commune, sont largement utilisés dans la technologie moderne.
Les cellules galvaniques sont utilisées comme sources d'énergie autonomes et de petite taille pour les machines, les appareils radio et les appareils de commande. Grâce à l'électrolyse, diverses substances sont obtenues, les surfaces sont traitées et des produits de la forme souhaitée sont créés.
Les processus électrochimiques ne profitent pas toujours aux humains et causent parfois de graves dommages, provoquant une corrosion accrue et la destruction des structures métalliques. Afin d’utiliser habilement les procédés électrochimiques et de lutter contre les phénomènes indésirables, il faut les étudier et pouvoir les réguler.
La cause des phénomènes électrochimiques est le transfert d'électrons ou un changement dans l'état d'oxydation des atomes de substances participant aux processus électrochimiques, c'est-à-dire des réactions redox se produisant dans des systèmes hétérogènes. Dans les réactions redox, les électrons sont directement transférés de l’agent réducteur à l’agent oxydant. Si les processus d'oxydation et de réduction sont spatialement séparés et que les électrons sont dirigés le long d'un conducteur métallique, alors un tel système représentera une cellule galvanique. La raison de l'apparition et du flux du courant électrique dans une cellule galvanique est la différence de potentiel.
Le potentiel de l'électrode. Potentiels des électrodes de mesure
Si vous prenez une plaque de n'importe quel métal et que vous la plongez dans l'eau, les ions de la couche superficielle, sous l'influence des molécules d'eau polaires, se détachent et s'hydratent dans le liquide. À la suite de cette transition, le liquide se charge positivement et le métal négativement, puisqu'un excès d'électrons y apparaît. L’accumulation d’ions métalliques dans le liquide commence à inhiber la dissolution du métal. Un équilibre mobile s’établit
Me 0 + mH 2 O = Me n + × m H 2 O + ne -
L'état d'équilibre dépend à la fois de l'activité du métal et de la concentration de ses ions en solution. Dans le cas des métaux actifs dans la série de tension jusqu'à l'hydrogène, l'interaction avec les molécules d'eau polaires se termine par la séparation des ions métalliques positifs de la surface et la transition des ions hydratés en solution (Fig. b). Le métal devient chargé négativement. Le processus est l’oxydation. À mesure que la concentration d’ions près de la surface augmente, le processus inverse devient possible : la réduction des ions. L’attraction électrostatique entre les cations en solution et les électrons en excès à la surface forme une double couche électrique. Cela conduit à l'apparition d'une certaine différence de potentiel, ou saut de potentiel, à l'interface entre le métal et le liquide. La différence de potentiel qui apparaît entre un métal et son environnement aqueux est appelée le potentiel de l'électrode. Lorsqu’un métal est immergé dans une solution d’un sel de ce métal, l’équilibre se déplace. L'augmentation de la concentration d'ions d'un métal donné dans la solution facilite le processus de transition des ions de la solution au métal. Les métaux dont les ions ont une capacité importante à passer en solution seront chargés positivement dans une telle solution, mais dans une moindre mesure que dans l'eau pure.
Pour les métaux inactifs, la concentration d’équilibre des ions métalliques en solution est très faible. Si un tel métal est immergé dans une solution d'un sel de ce métal, des ions chargés positivement sont libérés sur le métal à un rythme plus rapide que la transition des ions du métal à la solution. La surface métallique recevra une charge positive et la solution recevra une charge négative en raison de l'excès d'anions de sel. Et dans ce cas, une double couche électrique apparaît à l’interface métal-solution, d’où une certaine différence de potentiel (Fig. c). Dans le cas considéré, le potentiel de l'électrode est positif.
Riz. Le processus de transition d'un ion d'un métal à une solution :
un équilibre; b- dissolution ; c – dépôt
Le potentiel de chaque électrode dépend de la nature du métal, de la concentration de ses ions dans la solution et de la température. Si un métal est immergé dans une solution de son sel contenant une mole d'ion métallique pour 1 dm 3 (dont l'activité est de 1), alors le potentiel de l'électrode sera une valeur constante à une température de 25 o C et une pression de 1 au m. Ce potentiel est appelé potentiel d'électrode standard (Eo).
Les ions métalliques ayant une charge positive, pénétrant dans la solution et se déplaçant dans le champ potentiel de l'interface métal-solution, dépensent de l'énergie. Cette énergie est compensée par le travail de dilatation isotherme d’une concentration d’ions plus élevée en surface vers une concentration plus faible dans la solution. Les ions positifs s'accumulent dans la couche superficielle jusqu'à une concentration Avec Ô, puis passez en solution, où la concentration d'ions libres Avec. Le travail du champ électrique EnF est égal au travail de dilatation isotherme RTln(с o /с). En assimilant les deux expressions du travail, nous pouvons déduire l’ampleur du potentiel
En F = RTln(s o /s), -E = RTln(s/s o)/nF,
où E est le potentiel métallique, V ; R – constante universelle des gaz, J/mol K ; T – température, K ; n – charge ionique ; F – Numéro Faraday ; с – concentration d'ions libres ;
с о – concentration d'ions dans la couche superficielle.
Il n'est pas possible de mesurer directement la valeur du potentiel, car il est impossible de déterminer expérimentalement la valeur du potentiel. Les valeurs des potentiels d'électrode sont déterminées empiriquement par rapport à la valeur d'une autre électrode dont le potentiel est classiquement supposé nul. Une telle électrode étalon ou de référence est électrode à hydrogène normale (n.v.e.) . La structure de l'électrode à hydrogène est représentée sur la figure. Il se compose d'une plaque de platine recouverte de platine déposée électrolytiquement. L'électrode est immergée dans une solution 1M d'acide sulfurique (l'activité des ions hydrogène est de 1 mol/dm3) et est lavée par un courant d'hydrogène gazeux sous une pression de 101 kPa et T = 298 K. Lorsque le platine est saturé d'hydrogène , l'équilibre s'établit sur la surface métallique, le processus global est exprimé par l'équation
2Н + +2е ↔ Н 2 .
Si une plaque de métal immergée dans une solution 1M d'un sel de ce métal est reliée par un conducteur externe à une électrode à hydrogène standard, et que les solutions sont reliées par une clé électrolytique, alors on obtient une cellule galvanique (Fig. 32). La force électromotrice de cette cellule galvanique sera la quantité potentiel d'électrode standard d'un métal donné (E Ô ).

Schéma de mesure du potentiel d'électrode standard
par rapport à l'électrode à hydrogène
En prenant du zinc dans une solution 1 M de sulfate de zinc comme électrode et en le connectant avec une électrode à hydrogène, on obtient une cellule galvanique dont le circuit s'écrira comme suit :
(-) Zn/Zn 2+ // 2H + /H 2, Pt (+).
Dans le diagramme, une ligne indique l'interface entre l'électrode et la solution, deux lignes indiquent l'interface entre les solutions. L'anode est inscrite à gauche, la cathode à droite. Dans un tel élément, la réaction Zn o + 2H + = Zn 2+ + H 2 a lieu et les électrons traversent le circuit externe du zinc à l'électrode à hydrogène. Potentiel d'électrode standard pour électrode de zinc (-0,76 V).
En prenant une plaque de cuivre comme électrode, dans les conditions spécifiées en combinaison avec une électrode à hydrogène standard, on obtient une cellule galvanique
(-) Pt, H 2 /2H + //Cu 2+ /Cu (+).
Dans ce cas, la réaction se produit : Cu 2+ + H 2 = Cu o + 2H +. Les électrons se déplacent à travers le circuit externe de l'électrode à hydrogène vers l'électrode à cuivre. Potentiel d'électrode standard de l'électrode de cuivre (+0,34 V).
Si parmi toute la série de potentiels d'électrodes standard, nous sélectionnons uniquement les processus d'électrode qui correspondent à l'équation générale
nous obtenons alors une série de contraintes métalliques. En plus des métaux, cette série comprendra toujours l'hydrogène, ce qui vous permettra de voir quels métaux sont capables de déplacer l'hydrogène des solutions aqueuses d'acides.
Tableau 19. Séries de contraintes métalliques
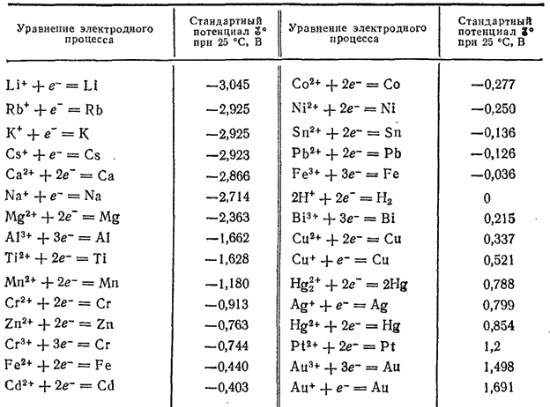
Un certain nombre de contraintes pour les métaux les plus importants sont indiquées dans le tableau. 19. La position d'un métal particulier dans la série de contraintes caractérise sa capacité à subir des interactions rédox dans des solutions aqueuses dans des conditions standard. Les ions métalliques sont des agents oxydants et les métaux sous forme de substances simples sont des agents réducteurs. De plus, plus un métal est situé loin dans la série de tension, plus l'agent oxydant dans une solution aqueuse sont ses ions, et vice versa, plus le métal est proche du début de la série, plus les propriétés réductrices d'un simple substance - le métal.
Potentiel du processus d'électrode
![]()
en milieu neutre, il est égal à B (voir page 273). Les métaux actifs en début de série, ayant un potentiel nettement supérieur à -0,41 V, déplacent l'hydrogène de l'eau. Le magnésium déplace l'hydrogène uniquement de l'eau chaude. Les métaux situés entre le magnésium et le cadmium ne déplacent généralement pas l'hydrogène de l'eau. Des films d'oxyde se forment à la surface de ces métaux, qui ont un effet protecteur.
Les métaux situés entre le magnésium et l'hydrogène déplacent l'hydrogène des solutions acides. Dans le même temps, des films protecteurs se forment également à la surface de certains métaux, inhibant la réaction. Ainsi, le film d'oxyde sur l'aluminium rend ce métal stable non seulement dans l'eau, mais aussi dans les solutions de certains acides. Le plomb ne se dissout pas dans l'acide sulfurique à sa concentration inférieure, car le sel formé lorsque le plomb réagit avec l'acide sulfurique est insoluble et crée un film protecteur sur la surface métallique. Le phénomène d'inhibition profonde de l'oxydation du métal, dû à la présence de films protecteurs d'oxyde ou de sel à sa surface, est appelé passivité, et l'état du métal dans ce cas est appelé état passif.
Les métaux sont capables de se déplacer les uns les autres à partir des solutions salines. La direction de la réaction est déterminée par leur position relative dans la série de contraintes. Lorsqu'on considère des cas spécifiques de telles réactions, il ne faut pas oublier que les métaux actifs déplacent l'hydrogène non seulement de l'eau, mais également de toute solution aqueuse. Par conséquent, le déplacement mutuel des métaux des solutions de leurs sels ne se produit pratiquement que dans le cas de métaux situés dans la série après le magnésium.
Beketov fut le premier à étudier en détail le déplacement des métaux de leurs composés par d'autres métaux. Grâce à son travail, il a classé les métaux en fonction de leur activité chimique dans une série de déplacements, qui est le prototype d'une série de contraintes métalliques.
La position relative de certains métaux dans la série de contraintes et dans le tableau périodique ne correspond pas à première vue. Par exemple, selon sa position dans le tableau périodique, l'activité chimique du potassium doit être supérieure à celle du sodium et celle du sodium est supérieure à celle du lithium. Dans la série de tensions, le lithium est le plus actif et le potassium occupe une position intermédiaire entre le lithium et le sodium. Le zinc et le cuivre, selon leur position dans le tableau périodique, devraient avoir une activité chimique à peu près égale, mais dans la série de tensions, le zinc est localisé beaucoup plus tôt que le cuivre. La raison de ce type d’incohérence est la suivante.
Lors de la comparaison de métaux occupant l'une ou l'autre position dans le tableau périodique, l'énergie d'ionisation des atomes libres est prise comme mesure de leur activité chimique - capacité réductrice. En effet, lors d'un déplacement, par exemple, de haut en bas le long du sous-groupe principal du groupe I du système périodique, l'énergie d'ionisation des atomes diminue, ce qui est associé à une augmentation de leurs rayons (c'est-à-dire à une plus grande distance des électrons externes du noyau) et avec un blindage croissant de la charge positive du noyau par des couches électroniques intermédiaires (voir § 31). Par conséquent, les atomes de potassium présentent une plus grande activité chimique (ils ont des propriétés réductrices plus fortes) que les atomes de sodium, et les atomes de sodium présentent une plus grande activité que les atomes de lithium.
Lors de la comparaison de métaux dans une série de tensions, le travail de conversion d'un métal à l'état solide en ions hydratés dans une solution aqueuse est considéré comme une mesure de l'activité chimique. Ce travail peut être représenté comme la somme de trois termes : l'énergie d'atomisation - la transformation d'un cristal métallique en atomes isolés, l'énergie d'ionisation des atomes métalliques libres et l'énergie d'hydratation des ions résultants. L'énergie d'atomisation caractérise la résistance du réseau cristallin d'un métal donné. L'énergie d'ionisation des atomes - l'élimination des électrons de valence - est directement déterminée par la position du métal dans le tableau périodique. L'énergie libérée lors de l'hydratation dépend de la structure électronique de l'ion, de sa charge et de son rayon.
Les ions lithium et potassium, ayant la même charge mais des rayons différents, créeront des champs électriques inégaux autour d’eux. Le champ généré à proximité des petits ions lithium sera plus fort que celui généré à proximité des gros ions potassium. Il en ressort clairement que les ions lithium s’hydrateront en libérant plus d’énergie que les ions potassium.
Ainsi, lors de la transformation considérée, de l'énergie est dépensée pour l'atomisation et l'ionisation et de l'énergie est libérée lors de l'hydratation. Plus la consommation totale d'énergie est faible, plus l'ensemble du processus sera facile et plus le métal donné sera proche du début de la série de contraintes. Mais sur les trois termes du bilan énergétique général, un seul - l'énergie d'ionisation - est directement déterminé par la position du métal dans le tableau périodique. Par conséquent, il n’y a aucune raison de s’attendre à ce que la position relative de certains métaux dans la série de contraintes corresponde toujours à leur position dans le tableau périodique. Ainsi, pour le lithium, la consommation totale d'énergie s'avère moindre que pour le potassium, selon lequel le lithium précède le potassium dans la série de tension.
Pour le cuivre et le zinc, la dépense énergétique pour l'ionisation des atomes libres et le gain énergétique lors de l'hydratation des ions sont proches. Mais le cuivre métallique forme un réseau cristallin plus résistant que le zinc, comme le montre la comparaison des températures de fusion de ces métaux : le zinc fond à , et le cuivre seulement à . Par conséquent, l'énergie dépensée pour l'atomisation de ces métaux est très différente, de sorte que les coûts énergétiques totaux pour l'ensemble du processus dans le cas du cuivre sont beaucoup plus élevés que dans le cas du zinc, ce qui explique la position relative de ces métaux. métaux dans la série de contraintes.
Lors du passage de l'eau à des solvants non aqueux, les positions relatives des métaux dans la série de tensions peuvent changer. La raison en est que l’énergie de solvatation des différents ions métalliques change différemment lorsqu’on passe d’un solvant à un autre.
En particulier, l'ion cuivre est solvaté assez vigoureusement dans certains solvants organiques ; Cela conduit au fait que dans de tels solvants, le cuivre se situe dans la série de tensions avant l'hydrogène et le déplace des solutions acides.
Ainsi, contrairement au système périodique d'éléments, une série de contraintes métalliques ne reflète pas un modèle général, sur la base duquel il est possible de donner une caractéristique globale des propriétés chimiques des métaux. Une série de tensions caractérise uniquement la capacité redox du système électrochimique « métal - ion métallique » dans des conditions strictement définies : les valeurs qui y sont indiquées se réfèrent à une solution aqueuse, à la température et à la concentration unitaire (activité) d'ions métalliques.
Grosse E., Weissmantel H.
Chimie pour les curieux. Bases de chimie et expériences divertissantes.
Chapitre 3 (suite)PETIT COURS D'ÉLECTROCHIMIE DES METAUX
Nous connaissons déjà l'électrolyse de solutions de chlorures de métaux alcalins et la production de métaux à l'aide de matières fondues. Essayons maintenant, à l'aide de quelques expériences simples, d'étudier certaines des lois de l'électrochimie des solutions aqueuses et des cellules galvaniques, et de nous familiariser également avec la production de revêtements galvaniques protecteurs.Les méthodes électrochimiques sont utilisées dans la chimie analytique moderne et servent à déterminer les quantités les plus importantes de la chimie théorique.
Enfin, la corrosion des objets métalliques, qui cause de graves dommages à l'économie nationale, est dans la plupart des cas un processus électrochimique.
SÉRIE CONTRAINTE DES MÉTAUX
Le lien fondamental pour comprendre les processus électrochimiques est la série de tensions des métaux. Les métaux peuvent être classés dans une série qui commence par les métaux nobles chimiquement actifs et se termine par les métaux nobles les moins actifs :Li, Rb, K, Ba, Sr, Ca, Mg, Al, Be, Mn, Zn, Cr, Ga, Fe, Cd, Tl, Co, Ni, Sn, Pb, H, Sb, Bi, As, Cu, Hg, Ag, Pd, Pt, Au.
Il s’agit, selon les idées les plus récentes, d’une série de tensions pour les métaux les plus importants et l’hydrogène. Si les électrodes d'une cellule galvanique sont constituées de deux métaux consécutifs, une tension négative apparaîtra sur le matériau précédant la rangée.
Valeur de tension ( potentiel électrochimique) dépend de la position de l'élément dans la série de tension et des propriétés de l'électrolyte.
Nous établirons l'essence de la série de tensions à partir de plusieurs expériences simples, pour lesquelles nous aurons besoin d'une source de courant et d'instruments de mesure électriques. Dissoudre environ 10 g de sulfate de cuivre cristallin dans 100 ml d'eau et plonger une aiguille en acier ou un morceau de tôle de fer dans la solution. (Nous vous recommandons de nettoyer d'abord le fer jusqu'à ce qu'il brille avec du papier de verre fin.) Après un court instant, le fer sera recouvert d'une couche rougeâtre de cuivre libéré. Un fer plus actif déplace le cuivre de la solution, le fer se dissolvant sous forme d'ions et le cuivre étant libéré sous forme de métal. Le processus se poursuit tant que la solution est en contact avec le fer. Une fois que le cuivre recouvre toute la surface du fer, il s’arrête pratiquement. Dans ce cas, une couche de cuivre plutôt poreuse se forme, de sorte que les revêtements protecteurs ne peuvent pas être obtenus sans l'utilisation de courant.
Dans les expériences suivantes, nous plongerons de petites bandes de tôle de zinc et de plomb dans une solution de sulfate de cuivre. Au bout de 15 minutes, nous les sortons, les lavons et les examinons au microscope. Nous pouvons discerner de magnifiques motifs semblables à de la glace, qui, à la lumière réfléchie, sont de couleur rouge et sont constitués de cuivre libéré. Ici aussi, des métaux plus actifs ont converti le cuivre de l’état ionique à l’état métallique.
À son tour, le cuivre peut déplacer les métaux qui sont plus bas dans la série de tension, c'est-à-dire moins actifs. Appliquez quelques gouttes de solution de nitrate d'argent sur une fine bande de feuille de cuivre ou de fil de cuivre aplati (après avoir préalablement nettoyé la surface pour la faire briller). À l'œil nu, vous pouvez voir la couche noirâtre qui en résulte, qui, au microscope et en lumière réfléchie, ressemble à de fines aiguilles et à des motifs végétaux (appelés dendrites).
Pour isoler le zinc sans courant, il faut utiliser un métal plus actif. Hors métaux qui réagissent violemment avec l’eau, on retrouve le magnésium dans la série de tension au-dessus du zinc. Placez quelques gouttes de solution de sulfate de zinc sur un morceau de ruban de magnésium ou sur de fins copeaux d'électrons. On obtient une solution de sulfate de zinc en dissolvant un morceau de zinc dans de l'acide sulfurique dilué. Avec le sulfate de zinc, ajoutez quelques gouttes d'alcool dénaturé. Sur le magnésium, après un court laps de temps, on remarquera, notamment au microscope, du zinc libéré sous forme de fins cristaux.
En général, n'importe quel membre de la série de tension peut être déplacé de la solution, où il existe sous forme d'ion, et converti à l'état métallique. Cependant, en essayant toutes sortes de combinaisons, on peut être déçu. Il semblerait que si une bande d'aluminium est immergée dans des solutions de sels de cuivre, de fer, de plomb et de zinc, ces métaux devraient s'y libérer. Mais cela n’arrive pas. La raison de la défaillance ne réside pas dans une erreur dans la série de tension, mais repose sur une inhibition particulière de la réaction, qui dans ce cas est due à un mince film d'oxyde à la surface de l'aluminium. Dans de telles solutions, l'aluminium est dit passif.
REGARDONS DANS LES COULISSES
Pour formuler les lois des processus en cours, on peut se limiter à considérer les cations et exclure les anions, puisqu'ils ne participent pas eux-mêmes à la réaction. (Cependant, la vitesse de dépôt est affectée par le type d'anions.) Si, par souci de simplicité, nous supposons que les métaux précipités et dissous produisent des cations doublement chargés, alors nous pouvons écrire :Moi 1 + Moi 2 2+ = Moi 1 2+ + Moi 2
De plus, pour la première expérience Me 1 = Fe, Me 2 = Cu.
Ainsi, le processus consiste en l’échange de charges (électrons) entre les atomes et les ions des deux métaux. Si l'on considère séparément (comme réactions intermédiaires) la dissolution du fer ou la précipitation du cuivre, on obtient :
Fe = Fe 2+ + 2 e --
Сu 2+ + 2 e-- = Cu
Considérons maintenant le cas où un métal est immergé dans l'eau ou dans une solution saline, avec un cation dont l'échange est impossible du fait de sa position dans la série de contraintes. Malgré cela, le métal a tendance à se dissoudre sous forme d’ion. Dans ce cas, l'atome métallique cède deux électrons (si le métal est divalent), la surface du métal immergée dans la solution devient chargée négativement par rapport à la solution et une double couche électrique se forme à l'interface. Cette différence de potentiel empêche une dissolution ultérieure du métal, de sorte que le processus s'arrête rapidement.
Si deux métaux différents sont immergés dans une solution, ils se chargeront tous les deux, mais le moins actif sera un peu plus faible, car ses atomes sont moins enclins à perdre des électrons.
Connectons les deux métaux avec un conducteur. En raison de la différence de potentiel, un flux d’électrons circulera du métal le plus actif vers le métal le moins actif, qui forme le pôle positif de l’élément. Un processus se produit dans lequel le métal le plus actif entre en solution et les cations de la solution sont libérés sur le métal le plus noble. Illustrons maintenant par quelques expériences le raisonnement quelque peu abstrait exposé ci-dessus (qui représente d'ailleurs une grossière simplification).
Tout d'abord, remplissez un bécher de 250 ml jusqu'au milieu avec une solution à 10 % d'acide sulfurique et plongez-y des morceaux de zinc et de cuivre pas trop petits. Nous soudons ou rivetons du fil de cuivre aux deux électrodes, dont les extrémités ne doivent pas toucher la solution.
Tant que les extrémités du fil ne sont pas reliées entre elles, on observera la dissolution du zinc, qui s'accompagne d'un dégagement d'hydrogène. Le zinc, comme le montre la série de tensions, est plus actif que l'hydrogène, de sorte que le métal peut déplacer l'hydrogène de l'état ionique. Une double couche électrique est formée sur les deux métaux. Le moyen le plus simple de détecter la différence de potentiel entre les électrodes consiste à utiliser un voltmètre. Immédiatement après avoir connecté l'appareil au circuit, la flèche indiquera environ 1 V, mais la tension chutera ensuite rapidement. Si vous connectez une petite ampoule qui consomme 1 V à l'élément, elle s'allumera - d'abord assez fortement, puis la lueur deviendra faible.
Sur la base de la polarité des bornes de l'appareil, nous pouvons conclure que l'électrode de cuivre est le pôle positif. Cela peut être prouvé sans appareil en considérant l’électrochimie du processus. Préparons une solution saturée de sel de table dans un petit bécher ou un tube à essai, ajoutons environ 0,5 ml d'une solution alcoolique de l'indicateur de phénolphtaléine et plongeons les deux électrodes fermées par du fil dans la solution. Une légère couleur rougeâtre sera observée près du pôle négatif, provoquée par la formation d’hydroxyde de sodium à la cathode.
Dans d’autres expériences, on peut placer différentes paires de métaux dans une cellule et déterminer la tension résultante. Par exemple, le magnésium et l'argent donneront une différence de potentiel particulièrement grande en raison de la distance importante qui les sépare d'une série de tensions, tandis que le zinc et le fer, au contraire, en donneront une très petite, inférieure à un dixième de volt. En utilisant de l’aluminium, nous ne recevrons pratiquement aucun courant grâce à la passivation.
Tous ces éléments, ou, comme disent les électrochimistes, ces circuits, présentent l'inconvénient que lors de la mesure du courant, la tension à leurs bornes chute très rapidement. Par conséquent, les électrochimistes mesurent toujours la valeur réelle de la tension dans un état hors tension en utilisant la méthode de compensation de tension, c'est-à-dire en la comparant à la tension d'une autre source de courant.
Considérons les processus dans l'élément cuivre-zinc plus en détail. A la cathode, le zinc entre en solution selon l'équation suivante :
Zn = Zn 2+ + 2 e --
Les ions hydrogène de l'acide sulfurique sont évacués au niveau de l'anode en cuivre. Ils attachent les électrons traversant le fil depuis la cathode de zinc et, par conséquent, des bulles d'hydrogène se forment :
2H + + 2 e-- = N2
Après un court laps de temps, le cuivre sera recouvert d’une fine couche de bulles d’hydrogène. Dans ce cas, l'électrode de cuivre se transformera en électrode d'hydrogène et la différence de potentiel diminuera. Ce processus est appelé polarisation des électrodes. La polarisation de l'électrode de cuivre peut être éliminée en ajoutant un peu de solution de bichromate de potassium à la cellule après la chute de tension. Après cela, la tension augmentera à nouveau, car le bichromate de potassium oxydera l'hydrogène en eau. Le bichromate de potassium agit dans ce cas comme un dépolarisant.
En pratique, on utilise des circuits galvaniques dont les électrodes ne sont pas polarisées, ou des circuits dont la polarisation peut être supprimée par l'ajout de dépolariseurs.
À titre d'exemple d'élément non polarisable, considérons l'élément Daniel, qui était souvent utilisé dans le passé comme source de courant. Il s'agit également d'un élément cuivre-zinc, mais les deux métaux sont immergés dans des solutions différentes. L'électrode de zinc est placée dans une cellule d'argile poreuse remplie d'acide sulfurique dilué (environ 20 %). La cellule d'argile est suspendue dans un grand verre contenant une solution concentrée de sulfate de cuivre, et au fond se trouve une couche de cristaux de sulfate de cuivre. La deuxième électrode de ce récipient est un cylindre en feuille de cuivre.
Cet élément peut être réalisé à partir d'un bocal en verre, d'une cellule en argile disponible dans le commerce (en dernier recours, on utilise un pot de fleur en fermant le trou au fond) et de deux électrodes de taille adaptée.
Pendant le fonctionnement de l'élément, le zinc se dissout pour former du sulfate de zinc et des ions de cuivre sont libérés au niveau de l'électrode de cuivre. Mais en même temps, l'électrode de cuivre n'est pas polarisée et l'élément produit une tension d'environ 1 V. En fait, théoriquement, la tension aux bornes est de 1,10 V, mais lors de la collecte de courant, nous mesurons une valeur légèrement inférieure en raison de la tension électrique. résistance de la cellule.
Si nous ne supprimons pas le courant de l’élément, nous devons retirer l’électrode de zinc de la solution d’acide sulfurique, sinon elle se dissoudra pour former de l’hydrogène.
Un schéma d'une cellule simple ne nécessitant pas de cloison poreuse est présenté sur la figure. L'électrode de zinc est située en haut du bocal en verre et l'électrode de cuivre est située près du fond. La cellule entière est remplie d'une solution saturée de sel de table. Placez une poignée de cristaux de sulfate de cuivre au fond du pot. La solution concentrée de sulfate de cuivre résultante se mélangera très lentement à la solution de sel de table. Ainsi, lorsque la cellule fonctionnera, du cuivre sera libéré sur l'électrode de cuivre, et le zinc se dissoudra sous forme de sulfate ou de chlorure dans la partie supérieure de la cellule.
De nos jours, les batteries utilisent presque exclusivement des piles sèches, plus pratiques à utiliser. Leur ancêtre est l'élément Leclanche. Les électrodes sont un cylindre de zinc et une tige de carbone. L'électrolyte est une pâte composée principalement de chlorure d'ammonium. Le zinc se dissout dans la pâte et de l'hydrogène se dégage sur le charbon. Pour éviter la polarisation, la tige de carbone est plongée dans un sac en lin contenant un mélange de poudre de charbon et de pyrolusite. La poudre de carbone augmente la surface de l'électrode et la pyrolusite agit comme un dépolarisant, oxydant lentement l'hydrogène.
Certes, la capacité dépolarisante de la pyrolusite est plus faible que celle du dichromate de potassium mentionné précédemment. Par conséquent, lorsque le courant est reçu dans des piles sèches, la tension chute rapidement, ils " se fatiguer"en raison de la polarisation. Ce n'est qu'après un certain temps que l'oxydation de l'hydrogène se produit avec la pyrolusite. Ainsi, les éléments " repos", si vous ne faites pas passer de courant pendant un certain temps. Vérifions cela sur une batterie pour lampe de poche, à laquelle nous connectons une ampoule. En parallèle avec la lampe, c'est-à-dire directement aux bornes, nous connectons un voltmètre.
Au début, la tension sera d'environ 4,5 V. (Le plus souvent, ces batteries ont trois cellules connectées en série, chacune avec une tension théorique de 1,48 V.) Après un certain temps, la tension chutera et la lueur de l'ampoule s'allumera. affaiblir. Sur la base des lectures du voltmètre, nous pouvons juger de la durée pendant laquelle la batterie doit se reposer.
Une place particulière est occupée par les éléments régénérants appelés batteries. Ils subissent des réactions réversibles et peuvent être rechargés après décharge de la cellule en se connectant à une source DC externe.
Actuellement, les batteries au plomb sont les plus courantes ; L'électrolyte qu'ils contiennent est de l'acide sulfurique dilué, dans lequel sont immergées deux plaques de plomb. L'électrode positive est recouverte de dioxyde de plomb PbO 2, la négative est en plomb métallique. La tension aux bornes est d'environ 2,1 V. Lors de la décharge, du sulfate de plomb se forme sur les deux plaques, qui se transforme à nouveau en plomb métallique et en peroxyde de plomb lors de la charge.
APPLICATION DE REVÊTEMENTS GALVANIQUES
Le dépôt de métaux à partir de solutions aqueuses à l'aide d'un courant électrique est le processus inverse de dissolution électrolytique, que nous avons connu en considérant les cellules galvaniques. Tout d’abord, nous examinerons le dépôt de cuivre, qui est utilisé dans un coulomètre à cuivre pour mesurer la quantité d’électricité.Le métal est déposé par le courant
Après avoir plié les extrémités de deux fines plaques de cuivre, nous les accrochons aux parois opposées d'un bécher ou, mieux encore, d'un petit aquarium en verre. Nous attachons les fils aux plaques avec des bornes.
Électrolyte Préparons selon la recette suivante : 125 g de sulfate de cuivre cristallin, 50 g d'acide sulfurique concentré et 50 g d'alcool (alcool dénaturé), le reste est de l'eau jusqu'à 1 litre. Pour ce faire, dissolvez d'abord le sulfate de cuivre dans 500 ml d'eau, puis ajoutez délicatement de l'acide sulfurique par petites portions ( Chauffage! Le liquide peut éclabousser !), puis ajoutez de l'alcool et ajoutez de l'eau jusqu'à un volume de 1 litre.
Remplissez le coulomètre avec la solution préparée et connectez une résistance variable, un ampèremètre et une batterie au plomb au circuit. À l'aide d'une résistance, nous ajustons le courant de manière à ce que sa densité soit de 0,02 à 0,01 A/cm 2 de surface de l'électrode. Si la plaque de cuivre a une superficie de 50 cm2, l'intensité du courant doit être comprise entre 0,5 et 1 A.
Après un certain temps, du cuivre métallique rouge clair commencera à précipiter à la cathode (électrode négative) et le cuivre entrera en solution à l'anode (électrode positive). Pour nettoyer les plaques de cuivre, nous ferons passer du courant dans le coulomètre pendant environ une demi-heure. Ensuite, nous retirons la cathode, la séchons soigneusement avec du papier filtre et la pesons avec précision. Installons une électrode dans la cellule, fermons le circuit à l'aide d'un rhéostat et maintenons un courant constant, par exemple 1 A. Au bout d'une heure, ouvrons le circuit et pesons à nouveau la cathode séchée. A un courant de 1 A, sa masse augmentera de 1,18 g par heure de fonctionnement.
Ainsi, une quantité d’électricité égale à 1 ampère-heure traversant une solution peut libérer 1,18 g de cuivre. Ou en général : la quantité de substance libérée est directement proportionnelle à la quantité d'électricité traversant la solution.
Pour isoler 1 équivalent d'un ion, il faut faire passer dans la solution une quantité d'électricité égale au produit de la charge de l'électrode e et du nombre d'Avogadro N UN:
e*N A = 1,6021 * 10 -19 * 6,0225 * 10 23 = 9,65 * 10 4 A * s * mol -1 Cette valeur est indiquée par le symbole F et porte le nom du découvreur des lois quantitatives de l'électrolyse Numéro Faraday(valeur exacte F- 96 498 A*s*mol -1). Par conséquent, pour isoler un nombre donné d’équivalents d’une solution n e une quantité d'électricité doit traverser la solution égale à F*n e A*s*mol -1 . Autrement dit,
Il =F*n euh ici je- actuel, t- temps de passage du courant à travers la solution. Au chapitre " Bases du titrage"Il a déjà été démontré que le nombre d'équivalents d'une substance n e est égal au produit du nombre de taupes et du nombre équivalent :
n e = n*Z Ainsi:
je*t = F*n*Z
Dans ce cas Z- charge ionique (pour Ag + Z= 1, pour Cu 2+ Z= 2, pour Al 3+ Z= 3, etc.). Si nous exprimons le nombre de moles comme le rapport entre la masse et la masse molaire ( n = m/m), on obtient alors une formule qui permet de calculer tous les processus se produisant lors de l'électrolyse :
Il =F*m*Z/M
En utilisant cette formule, vous pouvez calculer le courant :
je = F*m*Z/(t*M)= 9,65*10 4 *1,18*2 / (3600*63,54) A*s*g*mol/(s*mol*g) = 0,996 A
Si nous introduisons la relation pour le travail électrique W el
W el = U*I*t Et W e-mail/ U = Il
Puis, connaissant la tension U, vous pouvez calculer :
W el = F*m*Z*U/M
Il est également possible de calculer combien de temps il faut pour qu'une certaine quantité d'une substance soit libérée par électrolyse, ou quelle quantité d'une substance sera libérée dans un certain temps. Pendant l'expérience, la densité de courant doit être maintenue dans des limites spécifiées. Si elle est inférieure à 0,01 A/cm2, trop peu de métal sera libéré, car des ions cuivre(I) seront partiellement formés. Si la densité de courant est trop élevée, l'adhérence du revêtement à l'électrode sera faible et lorsque l'électrode sera retirée de la solution, elle risque de s'effriter.
Dans la pratique, les revêtements galvaniques sur les métaux sont principalement utilisés pour la protection contre la corrosion et pour obtenir un éclat miroir.
De plus, les métaux, en particulier le cuivre et le plomb, sont purifiés par dissolution anodique et séparation ultérieure à la cathode (raffinage électrolytique).
Pour plaquer du fer avec du cuivre ou du nickel, vous devez d’abord nettoyer soigneusement la surface de l’objet. Pour ce faire, polissez-le avec de la craie lavée et dégraissez-le successivement avec une solution diluée de soude caustique, d'eau et d'alcool. Si l'article est recouvert de rouille, vous devez le décaper au préalable dans une solution à 10-15 % d'acide sulfurique.
On suspend le produit nettoyé dans un bain électrolytique (un petit aquarium ou un bécher), où il servira de cathode.
La solution pour appliquer le cuivrage contient 250 g de sulfate de cuivre et 80 à 100 g d'acide sulfurique concentré dans 1 litre d'eau (Attention !). Dans ce cas, la plaque de cuivre servira d’anode. La surface de l'anode doit être approximativement égale à la surface de l'objet à revêtir. Il faut donc toujours veiller à ce que l’anode en cuivre soit suspendue dans le bain à la même profondeur que la cathode.
Le processus sera effectué à une tension de 3-4 V (deux batteries) et une densité de courant de 0,02-0,4 A/cm 2. La température de la solution dans le bain doit être comprise entre 18 et 25 °C.
Faisons attention au fait que le plan anodique et la surface à revêtir sont parallèles entre eux. Il vaut mieux ne pas utiliser d’objets aux formes complexes. En faisant varier la durée de l'électrolyse, il est possible d'obtenir des revêtements de cuivre de différentes épaisseurs.
Ils ont souvent recours à un cuivrage préalable afin d'appliquer un revêtement durable d'un autre métal sur cette couche. Ceci est particulièrement souvent utilisé lors du chromage du fer, du nickelage du zinc et dans d'autres cas. Il est vrai que des électrolytes cyanurés très toxiques sont utilisés à cette fin.
Pour préparer un électrolyte pour le nickelage, dissolvez 25 g de sulfate de nickel cristallin, 10 g d'acide borique ou 10 g de citrate de sodium dans 450 ml d'eau. Vous pouvez préparer vous-même du citrate de sodium en neutralisant une solution de 10 g d'acide citrique avec une solution diluée d'hydroxyde de sodium ou une solution de soude. Laissez l'anode être une plaque de nickel de la plus grande surface possible et prenez la batterie comme source de tension.
En utilisant une résistance variable, nous maintiendrons la densité de courant égale à 0,005 A/cm 2 . Par exemple, avec une surface d'objet de 20 cm 2, il faut travailler à une intensité de courant de 0,1 A. Après une demi-heure de travail, l'objet sera déjà nickelé. Sortons-le du bain et essuyons-le avec un chiffon. Il est toutefois préférable de ne pas interrompre le processus de nickelage, car la couche de nickel pourrait alors devenir passivée et le revêtement de nickel ultérieur n'adhérerait pas bien.
Pour obtenir un éclat miroir sans polissage mécanique, nous introduisons dans le bain galvanique un additif dit formateur de brillance. De tels additifs comprennent, par exemple, la colle, la gélatine et le sucre. Vous pouvez par exemple ajouter quelques grammes de sucre dans un bain de nickel et étudier son effet.
Pour préparer un électrolyte pour le chromage du fer (après cuivrage préalable), dissolvez 40 g d'anhydride chromique CrO 3 (Attention ! Poison !) et exactement 0,5 g d'acide sulfurique (en aucun cas plus !) dans 100 ml d'eau. Le processus se produit à une densité de courant d'environ 0,1 A/cm 2 et une plaque de plomb est utilisée comme anode, dont la surface doit être légèrement inférieure à la surface de la surface chromée.
Il est préférable de chauffer légèrement les bains de nickel et de chrome (jusqu'à environ 35°C). Veuillez noter que les électrolytes pour le chromage, notamment lors d'un processus long et à courant élevé, émettent des vapeurs contenant de l'acide chromique, très nocives pour la santé. Le chromage doit donc être réalisé sous traction ou à l’air libre, par exemple sur un balcon.
Lors du chromage (et dans une moindre mesure du nickelage), la totalité du courant n’est pas utilisée pour le dépôt du métal. Dans le même temps, de l’hydrogène est libéré. Sur la base d'un certain nombre de tensions, on pourrait s'attendre à ce que les métaux situés devant l'hydrogène ne soient pas du tout libérés des solutions aqueuses, mais qu'au contraire, moins d'hydrogène actif soit libéré. Cependant, ici, comme dans le cas de la dissolution anodique des métaux, le dégagement cathodique d'hydrogène est souvent inhibé et n'est observé qu'à haute tension. Ce phénomène est appelé surtension d'hydrogène et il est particulièrement important, par exemple pour le plomb. Grâce à cette circonstance, une batterie au plomb peut fonctionner. Lors du chargement d'une batterie, au lieu de PbO 2, de l'hydrogène devrait apparaître à la cathode, mais, en raison d'une surtension, le dégagement d'hydrogène commence lorsque la batterie est presque complètement chargée.
Corrosion électrochimique du métal. La protection cathodique. Protection anodique. Protection passive. Potentiels d'électrodes - tableau.
Dans la grande majorité des cas, la corrosion des métaux fait référence à l’oxydation d’un matériau. Dans la pratique, le plus grand mal est causé par ce qu'on appelle. corrosion électrochimique accompagnée d'un transfert actif de matière. Les surfaces métalliques sont sensibles à la destruction électrochimique (corrosion) lorsqu'elles entrent en contact avec des électrolytes (agents de corrosion). Ces agents peuvent être des gaz atmosphériques, tels que l'air marin, urbain ou industriel (c'est-à-dire le dioxyde de soufre, le chlorure d'hydrogène et les sulfites, etc.) ou des liquides actifs - saumures, alcalis, eau de mer, etc. (par exemple, des empreintes de mains moites).
Si un couple galvanique se forme à la suite du contact d'un agent de corrosion sur des surfaces métalliques, le transfert d'une substance d'une électrode du couple à l'autre est plusieurs fois intensifié. Le taux de corrosion est déterminé par la différence des potentiels d'électrode de la paire. Ce processus est généralement entendu lorsqu'on parle de corrosion électrochimique.
Ayant tendance à abandonner des électrons, en raison du potentiel négatif de l’électrode, la plupart des métaux s’oxydent au cours du processus de corrosion. Si un certain potentiel positif supplémentaire est appliqué à l'objet protégé = un certain potentiel négatif de l'ordre du dixième de volt y est maintenu, alors la probabilité d'une réaction d'oxydation tombe presque à zéro. Cette méthode de protection est généralement sous-entendue lorsqu'on parle de la protection cathodique.
Si une certaine quantité d'une substance avec un potentiel d'électrode inférieur (par exemple, du zinc ou du magnésium pour protéger le fer) est placée au point de corrosion probable, alors une réaction d'oxydation se produira dessus. Un bon contact électrique doit être assuré entre cet élément supplémentaire anode de protection(anode sacrificielle) et métal protégé. Avez-vous deviné pourquoi les tuyaux sont galvanisés ? Qu’en est-il des tôles pour la toiture ? Naturellement, lorsque l’anode protectrice se dissoudra entièrement, tout se passera comme d’habitude.
Sous protection passive comprendre le revêtement de l'échantillon protégé avec un diélectrique pour éviter l'apparition d'un circuit galvanique. Par exemple, vous pouvez peindre une structure métallique avec de la peinture à l'huile, etc.
Tableau. Potentiels d'électrode standard de certaines substances :
| Matériel | Potentiel en V | Métal (M) Pas de métal (NM) |
| Lithium (Li) | -3.04 | M |
| Potassium (K) | -2.92 | M |
| Baryum (Ba) | -2.90 | M |
| Calcium (Ca) | -2.87 | M |
| Sodium (Na) | -2.71 | M |
| Magnésium (Mg) | -2.36 - -2.37 | M |
| Aluminium (Al) | -1.68 | M |
| Manganèse (Mn) | -1.18 - -1.19 | M |
| Zinc (Zn) | -0.76 | M |
| Chrome (Cr) | -0.74 | M |
| Soufre (S), solide | -0.48 - -0.51 | Nouveau-Mexique |
| Fer (Fe) | -0.41 - -0.44 | M |
| Cadmium (Cd) | -0.40 | M |
| Thallium (Tl) | -0.34 | M |
| Cobalt (Co) | -0.28 | M |
| Nickel (Ni) | -0.23 | M |
| Étain (Sn) | -0.14 | M |
| Plomb (Pb) | -0.13 | M |
| Hydrogène (2H) | 0.00 | |
| Cuivre (Cu) | +0.15 | M |
| Iode (I), solide | +0.54 | Nouveau-Mexique |
| Argent (Ag) | +0.80 | M |
| Mercure (Hg) | +0.85 | M |
| Brome (Br), liquide | +1.07 | Nouveau-Mexique |
| Platine (Pt) | +1.20 | M |
| Chlore (Cl), gaz | +1.36 | Nouveau-Mexique |
| Or (Au) | +1.50 | M |
| Fluor (F), gaz | +2.87 | Nouveau-Mexique |







