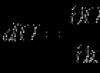PRÉVISION DU VOLUME CRITIQUE
où v sont des contributions partielles dont les valeurs, exprimées en cm3/mol, sont données dans le tableau. 5.2. Le calcul est assez simple et ne nécessite aucun commentaire supplémentaire.
PRÉDICTION DU FACTEUR ACENTRIQUE
Facteur acentrique
a été proposé en 1955 par Pitzer comme paramètre de corrélation caractérisant l'acentricité, ou non-sphéricité, d'une molécule. En analysant la dépendance de la pression réduite de vapeur saturée de diverses substances sur la température réduite, Pitzer et ses collègues ont découvert que pour l'argon, le krypton, le xénon, l'azote, l'oxygène, le monoxyde de carbone, le méthane et certaines autres substances, cette dépendance est décrite par presque une équation. Cependant, l'élargissement de cette liste avec des composés d'autres classes produit une série de lignes presque droites dont les pentes varient. Pitzer et al. ont adopté la pression de vapeur réduite. ![]() à une certaine température réduite
à une certaine température réduite ![]() comme caractéristique d'une substance. A ces températures, la pression réduite des gaz rares choisis comme substance simple est d'environ 0,1. Sur la base de cette observation, une définition d'un nouveau paramètre a été formulée : le facteur acentrique
comme décrivant l'écart de la valeur de la pression de vapeur réduite pour une certaine substance par rapport à la pression de vapeur réduite de la substance de comparaison sous la forme suivante :
comme caractéristique d'une substance. A ces températures, la pression réduite des gaz rares choisis comme substance simple est d'environ 0,1. Sur la base de cette observation, une définition d'un nouveau paramètre a été formulée : le facteur acentrique
comme décrivant l'écart de la valeur de la pression de vapeur réduite pour une certaine substance par rapport à la pression de vapeur réduite de la substance de comparaison sous la forme suivante :
(à Tr =0,7),(5.18)
où est la pression de vapeur saturée de la substance à la température donnée Tr =0,7.
Selon la définition de Pitzer, le facteur acentrique est « une mesure de l’écart des fonctions du potentiel intermoléculaire par rapport aux fonctions du potentiel intermoléculaire des molécules sphériques de la substance de référence ». Signification = 0 correspond à une symétrie sphérique dans un gaz raréfié. Les écarts par rapport au comportement caractéristique d'une substance simple sont évidents si > 0. Pour les gaz monoatomiques, le facteur acentrique est proche de zéro. Pour le méthane, c’est encore très faible. Cependant, pour les hydrocarbures de poids moléculaire élevé, la valeur augmente et augmente fortement avec l'augmentation de la polarité des molécules.
La plage de variation du facteur acentrique va de zéro à un. Actuellement, le facteur acentrique est largement utilisé comme paramètre qui caractérise dans une certaine mesure la complexité de la structure d'une molécule en ce qui concerne à la fois sa géométrie et sa polarité. Il est recommandé que l'applicabilité des corrélations incluant un facteur d'acentricité soit limitée aux gaz et liquides normaux et ne soit pas utilisée pour prédire les propriétés de liquides hautement polaires ou associés.
Il convient de noter ici que l'expérience de nos travaux nous permet de conclure que la limitation ci-dessus est trop catégorique. Sous réserve de certaines conditions de corrélation avec peut également être utilisé en relation avec les groupes nommés de substances organiques.
Les valeurs du facteur acentrique pour de nombreuses substances sont calculées sur la base des meilleures données expérimentales sur les pressions de vapeur, Tc Et PC connexions et sont contenus dans l’Annexe.
En l'absence d'informations sur pour le prédire, on peut utiliser :
L'équation d'Edmister
 ;(5.19)
;(5.19)
· Équation de Lee-Kesler
Équation d'Ambrose-Walton
 ,(5.21)
,(5.21)
Où - pression critique exprimée en atmosphères physiques ;
= - point d'ébullition normal réduit de la substance ;
- point d'ébullition normal d'une substance en degrés Kelvin ;
- température critique en degrés Kelvin.
f (0) , f (1) – défini dans la description de la méthode Ambrose-Walton (section 7.3)
Pour conclure notre examen du matériau sur les propriétés critiques et les critères de similarité, attardons-nous sur une autre question importante et générale. Il s'agit de critères de similarité. Actuellement, il y en a beaucoup qui sont proposés, nous avons fait connaissance avec l'un d'eux - le facteur acentrique. Insecte. 7 considère un autre critère de similarité - le coefficient de Riedel. Les deux critères sont très largement utilisés. Néanmoins, des approches universelles pour la sélection de l'un ou l'autre critère de similarité n'ont pas encore été créées, ce qui signifie que les travaux dans ce sens se poursuivront. Nous jugeons approprié de répéter les exigences énumérées par Wales dans sa monographie et liées à des paramètres supplémentaires ou à des critères de similarité :
· Ces paramètres doivent être liés à la structure moléculaire et aux propriétés électrostatiques de la molécule.
· Ils peuvent être déterminés avec un minimum de données expérimentales.
· Les propriétés critiques ne doivent pas affecter directement leurs valeurs.
· Lors de l'estimation de ces paramètres, il convient d'éviter d'utiliser des données sur PVT, sinon le sens de l'équation ci-dessus est perdu.
Les paramètres supplémentaires doivent être fonction de la température, de préférence donnée.
Vous pouvez être d'accord ou en désaccord avec les exigences énumérées, mais il est bien évident que ni le facteur acentrique ni le critère de Riedel ne répondent à l'ensemble de leur complexe. De plus, il nous semble clair que l’une des raisons du succès de leur application est précisément la cohérence de leurs valeurs avec les paramètres critiques et les données P-T. Le support de la connexion avec les données P-T est la température d'ébullition à l'une des pressions, le plus souvent à la pression atmosphérique.
Ainsi, le développement de méthodes de prévision nécessitera probablement une clarification des exigences en matière de critères de similarité.
6. PRÉDICTION de la densité des gaz et des liquides
Avant de passer à la prédiction, il convient de rappeler que, selon la température et la pression adoptées, une substance peut être soit dans un état saturé, soit insaturé. La pression au-dessus d'un liquide saturé est égale à sa pression de vapeur saturée à une température donnée. La pression au-dessus d'un liquide insaturé, surfondu ou comprimé est supérieure à sa pression de vapeur saturée à la température choisie pour le calcul. Pour chacune des zones nommées PVT l’espace, il existe des approches indépendantes pour prédire la densité.
Prédire la densité de substances individuelles à l'aide du coefficient de compressibilité
Exemple 6.1
Pour l'isobutylbenzène, qui a une température critique de 650 K, une pression critique de 31 atm et un facteur acentrique de 0,378, calculez à l'aide des tableaux de Lee-Kesler (tableaux 4.6, 4.7) :
· coefficient de compressibilité à 500, 657 et 1170 K et pression 1-300 atm,
· densité à 500, 657 et 1170 K et pression 1-300 atm ;
donner des dépendances graphiques :
· coefficient de compressibilité en fonction de la pression à des températures spécifiées,
· densité en fonction de la pression à des températures spécifiées.
Solution
Nous utilisons le développement de Pitzer (équation 4.34) et le tableau. 4,6, 4,7 pour le coefficient de compressibilité.
1. Calculons les valeurs des températures données :
500/600 =0,769; = 657/650 =1,01; = 1170/650 =1,80.
2. Calculons les valeurs des pressions données :
1/31 =0,03226; = 300/31 =9,677.
Étant donné que la plage de pressions réduites d'intérêt coïncide avec la plage considérée par Lee-Kesler, nous utilisons des informations sur et pour les valeurs discrètes présentées dans le tableau. 4.6, 4.7.
Chacune des valeurs et est obtenue par interpolation linéaire par rapport à la température. Donc, à 500 K (= 0,769) et = 0,010 car on a
(0,9935-0,9922)/(0,80-0,75)·(0,769-0,75)+0,9922 = 0,9927.
Prédire la densité des liquides et des vapeurs saturés à l'aide d'équations d'état de la matière
Trouver des conditions de saturation à partir des équations d'état est une tâche assez complexe, dont la solution est souvent impossible sans l'intervention de technologies informatiques et de logiciels spéciaux. Pour les équations d’état simples, comme l’équation de van der Waals, ce problème peut être résolu par des calculs simples. Cependant, il faut rappeler qu'en pratique, à l'aide de l'équation de Van der Waals, on ne peut évaluer que qualitativement l'état de saturation. Pour représenter plus précisément la saturation, d'autres équations d'état et méthodes spéciales ont été développées.
Dans ce manuel, en utilisant l'équation de van der Waals comme exemple, nous envisageons une approche pour trouver la pression de saturation et les volumes de saturation de liquide et de vapeur (points appartenant au binodal), ainsi que les conditions qui déterminent les états métastables de la matière. (points extrêmes isothermes).
Exemple 6.3
Pour l'isobutylbenzène à des températures de 400, 500, 600 et 640 K, à l'aide de l'équation de van der Waals, calculez la pression de vapeur et les volumes de saturation du liquide et de la vapeur. Déterminez également les régions d’états métastables de vapeur et de liquide aux températures indiquées. La température critique est de 650 K, la pression critique est de 31 atm.
Solution
1. Écrivons le principe de Maxwell :
Superficie = ![]() .(6.1)
.(6.1)
Exprimons la valeur de pression de l'équation de van der Waals et substituons-la dans l'intégrande. Nous obtenons
 .
(6.2)
.
(6.2)
Dans ce cas, il est possible de trouver une solution analytique à une certaine intégrale
 .(6.3)
.(6.3)
La tâche consiste maintenant à trouver la valeur de P assis, à laquelle l'expression 6.3 devient identique. Pour le trouver, nous devrons déterminer à plusieurs reprises les valeurs des volumes de liquide et de vapeur pour un P donné, c'est-à-dire trouver des solutions (racines) de l'équation cubique.
2. Réécrivons l'équation de van der Waals sous forme de polynôme en volume
 .(6.4)
.(6.4)
Les racines de cette équation peuvent être trouvées à l’aide des formules de Cardano. Pour ce faire, passons à la forme réduite de l'équation cubique en effectuant les transformations suivantes. Notons les coefficients de l'équation (6.4) par
 ; ;
; ;
et remplacez l'inconnu V par Y :
alors l'équation (6.4) prendra la forme réduite
![]() ,(6.5)
,(6.5)
Où ;  .
.
Le nombre de solutions réelles d'une équation cubique dépend du signe du discriminant
![]() .(6.6)
.(6.6)
Si D > 0, alors l'équation a une solution valide ; si D< 0, то - три действительных решения; и если D = 0, то уравнение имеет либо два действительных решения, одно из которых двукратное, либо одно действительное трехкратное решение (последнее в случае p = q = 0).
Cet exemple considère la région de l'espace PVT où coexistent vapeur et liquide. Pour cette région, l'équation de van der Waals a trois solutions réelles (le discriminant de l'équation (6.5) est inférieur à zéro). Lorsque l'on utilise les formules de Cardano dans leur forme originale, les racines de l'équation sont exprimées par des quantités complexes. Ceci peut être évité en introduisant la notation suivante :
![]() ,
, ![]() .(6.7)
.(6.7)
Alors les solutions de l’équation donnée (6.5) seront
![]() ;(6.8)
;(6.8)
de quel remplacement
![]() (6.11)
(6.11)
encore une fois, nous pouvons passer aux solutions de l'équation cubique (6.4).
3. Calculons les constantes caractéristiques de l'équation de van der Waals. Pour faciliter les calculs, nous accepterons les unités de mesure suivantes : V - l/mol, P - atm, T - K. Alors R = 0,08206 l atm/(mol K) ;
a = 27·0,082062·6502/(64·31)=38,72 l·atm ;
b = 0,08206,650/(8,31)=0,2151 l.
4. La pression de saturation est déterminée par la méthode des approximations successives. En première approximation à T = 400 K, on prend la pression de saturation égale à 10 atm.
5. Calculez les valeurs des coefficients de l'équation (6.4) :
 = –(0,2151+0,08206·400/10) = – 3,4975 ;
= –(0,2151+0,08206·400/10) = – 3,4975 ;
38,72/10 = 3,872;
= – (38,72·0,2151/10) = – 0,8329.
= /3 = – 0,2055;
 = 2·(–3,4975)3/27–(–3,4975·3,872)/3+(–0,8329)=0,5121 ;
= 2·(–3,4975)3/27–(–3,4975·3,872)/3+(–0,8329)=0,5121 ;
![]() = (–0,2055/3)3+(0,5121/2)2 = 0,0652.
= (–0,2055/3)3+(0,5121/2)2 = 0,0652.
La valeur discriminante (D) s'est avérée positive, ce qui indique la seule solution valable à l'équation (6.5). Par conséquent, la valeur de pression a été mal sélectionnée.
7. Supposons que la pression de saturation soit de 1 atm. Répétons les calculs des étapes 5 et 6.
 = –(0,2151+0,08206·400/1) = –33,04 ;
= –(0,2151+0,08206·400/1) = –33,04 ;
38,72/1 = 38,72;
= –(38,72·0,2151/1) = –8,329 ;
=/3 = –325,2;
 = 2·(–33,04)3/27 –(–33,04·38,72)/3+(–8,329) = –2254 ;
= 2·(–33,04)3/27 –(–33,04·38,72)/3+(–8,329) = –2254 ;
![]() = (–325,2/3)3+(–2254/2)2 = –3632.
= (–325,2/3)3+(–2254/2)2 = –3632.
8. Trouvons ces solutions, mais calculons d’abord les grandeurs auxiliaires et
![]() = [–(–325,2)3/27]1/2 = 1129;
= [–(–325,2)3/27]1/2 = 1129;
![]() = –(–2254)/(2·1129) = 0,9982 ;
= –(–2254)/(2·1129) = 0,9982 ;
= arccos (0,9982) = 0,0600 radians ;
![]() = 2·(1129)1/3·cos(0,0600/3) = 20,82 ;
= 2·(1129)1/3·cos(0,0600/3) = 20,82 ;
2·(1129)1/3 cos(0,0600/3 + 2·3,14/3) = –10,75 ;
2·(1129)1/3 cos (0,0600/3 + 4·3,14/3) = –10,09.
9. Passons aux solutions de l'équation (6.4), en utilisant (6.11).
![]() = 20,82 –(–33,04/3) = 31,8 l/mol ;
= 20,82 –(–33,04/3) = 31,8 l/mol ;
![]() = –10,75 –(–33,04/3) = 0,263 l/mol ;
= –10,75 –(–33,04/3) = 0,263 l/mol ;
![]() = –10,09 –(–33,04/3) = 0,923 l/mol.
= –10,09 –(–33,04/3) = 0,923 l/mol.
A 400 K et 1 atm, le volume de vapeur ( V1) est de 31,8 l/mol, le volume de liquide ( V2) – 0,263 l/mol. V3= 0,923 – la troisième racine de l'équation, qui n'a aucune signification physique.
10. Calculons la valeur du côté gauche de l'expression (6.3), pour cela nous avons toutes les quantités nécessaires :
 = 0,08206·400 ln[(31,8–0,2151)/
= 0,08206·400 ln[(31,8–0,2151)/
/(0,263-0,2151)] + 38,72·(1/31,8-1/0,263)–1·(31,8-0,263) = 35,53.
A la pression choisie (1 atm), l'expression (6.3) ne devient pas une identité, c'est-à-dire les côtés gauche et droit ne sont pas égaux. Une valeur de pression de saturation différente doit être adoptée.
Aux paragraphes 5 à 10, les calculs ont été effectués avec arrondi des valeurs intermédiaires à chaque étape des calculs aux valeursécrites dans les formules. Les calculs suivants sont présentés avec 16 décimales et l'arrondi est effectué uniquement lors de la présentation des valeurs finales.
11. Acceptons Psat= 3 guichets automatiques. Répétons les calculs des étapes 5 à 10. A 400 K et 3 atm, le volume de vapeur est de 9,878 l/mol, le volume de liquide est de 0,282 l/mol. Le côté gauche de l’expression (6.3) est égal à = 1,0515. L'identité n'est pas satisfaite, mais le degré d'écart par rapport à celle-ci a considérablement diminué.
12. La sélection de la pression de saturation doit être poursuivie. Il existe maintenant deux valeurs pour le côté gauche de l'expression (6.3) aux pressions correspondantes. En utilisant ces valeurs, vous pouvez estimer la valeur de pression pour le prochain calcul par interpolation linéaire.
= 1–(1–3)/(35,53–1,0515) 35,53 = 3,061 guichets automatiques.
13. Répétons les calculs (étapes 5 à 12) pour Psat= 3,061 guichets automatiques. On obtient :
= 9,658 l/mole ; = 0,282 l/mole ; = 0,473. La nouvelle valeur de pression est de 3,111 atm.
Après 5 itérations, hors calcul à Psat= 10 atm, on a :
T= 400K ; P. assis = 3,112 guichets automatiques ; = 9,480 l/mole ; = 0,282 l/mole ; = 8.7·10-5. Les valeurs obtenues de pression et de volumes de liquide et de vapeur correspondent aux conditions de saturation.
14. Les résultats des calculs pour d'autres températures sont donnés dans le tableau. 6.3.
Tableau 6.3
15. La région des états métastables (sursaturés) de la vapeur et du liquide occupe l'espace entre le binodal et le spinodal. Les points sur les isothermes appartenant au binodal sont définis ci-dessus, et leurs valeurs sont données dans le tableau. 6.3.
Pour déterminer la configuration spinodale, nous utilisons la relation
![]() ,
,
ceux. conditions d'extrémité pour les points isothermes correspondants. Ensuite, nous différencions l'équation de van der Waals par rapport au volume (à T = const) et transformons l'expression résultante en un polynôme dans V. Nous obtenons l'équation cubique (6.12), dont les racines peuvent être trouvées de la manière décrite ci-dessus (éléments 5 à 9) :
16. Pour 400 K on a les valeurs suivantes des coefficients de l'équation (6.12) :
= – = –2,3593;
1,0149;
= – = –0,1092.
Les coefficients de l'équation cubique réduite (6.5) sont respectivement égaux à :
= /3 = –0,8405;
 = 2·(–2,3593)3/27 –(–2,3593·1,0149)/3 + (–0,1092) = –0,2838 ;
= 2·(–2,3593)3/27 –(–2,3593·1,0149)/3 + (–0,1092) = –0,2838 ;
![]() = (–0,8405/3)3 + (–0,2838/2)2 = –0,0019.
= (–0,8405/3)3 + (–0,2838/2)2 = –0,0019.
La valeur de D est négative, donc l’équation a trois solutions réelles.
17. Trouvons les valeurs des racines de l'équation (6.12) à 400 K. Pour ce faire, nous effectuons séquentiellement les calculs suivants :
![]() = [–(–0,8405)3/27]1/2 = 0,1483;
= [–(–0,8405)3/27]1/2 = 0,1483;
![]() = –(–0,2838)/(2·0,1483) = 0,9568 ;
= –(–0,2838)/(2·0,1483) = 0,9568 ;
= arccos (0,9568) = 0,2950 radians ;
![]() = 2·(0,1483)1/3 cos(0,2950/3) = 1,0535 ;
= 2·(0,1483)1/3 cos(0,2950/3) = 1,0535 ;
2·(0,1483)1/3 cos(0,2950/3 + 2·3,14/3) = –0,6159 ;
2·(0,1483)1/3 cos(0,2950/3 + 4·3,14/3) = –0,4388 ;
![]() = 1,0535 –(–2,3593/3) = 1,840 l/mol ;
= 1,0535 –(–2,3593/3) = 1,840 l/mol ;
![]() = –0,6159 –(–2,3593/3) = 0,171 l/mol ;
= –0,6159 –(–2,3593/3) = 0,171 l/mol ;
![]() = –0,4388 –(–2,3593/3) = 0,348 l/mol.
= –0,4388 –(–2,3593/3) = 0,348 l/mol.
La plus grande racine = 1,840 l/mol correspond au maximum sur l'isotherme 400 K et limite les états métastables de la vapeur à gauche. La racine égale à 0,171 l/mol n'a pas d'interprétation physique, puisque sa valeur est inférieure au paramètre b de l'équation de van der Waals. Et enfin, la racine correspond au minimum sur l'isotherme de 400 K et sépare la région du liquide sursaturé des états absolument instables à gauche.
18. La pression dans le système avec le volume correspondant de vapeur sursaturée () et de liquide sursaturé () est trouvée à partir de l'équation de van der Waals en y substituant les valeurs de température et de volume requises.
 = (0,08206·400)/(1,840–0,215)–38,72/1,8402 = 8,763 guichet automatique ;
= (0,08206·400)/(1,840–0,215)–38,72/1,8402 = 8,763 guichet automatique ;
 = (0,08206·400)/(0,348–0,215)–38,72/0,3482 = –72,928 guichet automatique.
= (0,08206·400)/(0,348–0,215)–38,72/0,3482 = –72,928 guichet automatique.
19. Les résultats des calculs pour les autres températures sont donnés dans le tableau. 6.4.
De la théorie de Friedman, il résulte que différents scénarios d'évolution de l'Univers sont possibles : une expansion illimitée, une alternance de contractions et d'expansions, et même un état stationnaire trivial. Lequel de ces scénarios se réalisera dépend de la relation entre la densité critique et réelle de matière dans l'Univers à chaque étape de l'évolution. Afin d’estimer les valeurs de ces densités, considérons d’abord comment les astrophysiciens imaginent la structure de l’Univers.
On pense actuellement que la matière dans l’Univers existe sous trois formes : la matière ordinaire, le rayonnement cosmique de fond micro-ondes et la matière dite « noire ». La matière ordinaire est concentrée principalement dans les étoiles, qui sont au nombre d'environ cent milliards rien que dans notre Galaxie. La taille de notre Galaxie est de 15 kiloparsecs (1 parsec = 30,8 X 1012 km). On suppose qu'il existe jusqu'à un milliard de galaxies différentes dans l'Univers, dont la distance moyenne est de l'ordre d'un mégaparsec. Ces galaxies sont réparties de manière extrêmement inégale, formant des amas. Cependant, si nous considérons l'Univers à très grande échelle, par exemple en le « divisant » en « cellules » d'une taille linéaire supérieure à 300 mégaparsecs, alors la structure inégale de l'Univers ne sera plus observée. Ainsi, à très grande échelle, l’Univers est homogène et isotrope. Pour une distribution aussi uniforme de la matière, nous pouvons calculer la densité rв, qui est de ~ 3×10-31 g/cm3.
La densité équivalente au rayonnement de fond cosmique micro-ondes est rр ~ 5Х10-34 g/cm3, ce qui est bien inférieur à rв et peut donc ne pas être prise en compte lors du calcul de la densité totale de matière dans l'Univers.
En observant le comportement des galaxies, les scientifiques ont suggéré qu'en plus de la matière lumineuse et « visible » des galaxies elles-mêmes, dans l'espace qui les entoure, il existe apparemment des masses de matière importantes qui ne peuvent pas être observées directement. Ces masses « cachées » ne se manifestent que par la gravité, qui affecte le mouvement des galaxies en groupes et en amas. Sur la base de ces caractéristiques, on estime également la densité rt associée à cette matière « noire », qui, selon les calculs, devrait être environ 30 fois supérieure à rt. Comme on le verra dans ce qui suit, c'est la matière « noire » qui est finalement « responsable » de l'un ou l'autre « scénario » de l'évolution de l'Univers 1.
Pour le vérifier, estimons la densité critique de matière, à partir de laquelle le scénario évolutif « pulsé » cède la place à un scénario « monotone ». Une telle estimation, bien que grossière, peut être réalisée sur la base de la mécanique classique, sans faire appel à la théorie de la relativité générale. De l'astrophysique moderne, nous n'avons besoin que de la loi de Hubble.
Calculons l'énergie d'une certaine galaxie de masse m, située à une distance L de « l'observateur » (Fig. 1.1). L'énergie E de cette galaxie est constituée de l'énergie cinétique T = mv2/2 = mH2L2/2 et de l'énergie potentielle U = - GMm / L, qui est associée à l'interaction gravitationnelle de la galaxie m avec la matière de masse M située à l'intérieur d'une boule de rayon L (on peut montrer que la matière, située à l'extérieur de la boule, ne contribue pas à l'énergie potentielle). En exprimant la masse M par la densité r, M = 4pL3r/3, et en tenant compte de la loi de Hubble, on écrit l'expression de l'énergie de la galaxie :
E = T - G 4/3 pmr v2/H2 = T (1-G 8pr/3H2) (1.1).
Figure 1.1.
De cette expression il ressort clairement que, selon la valeur de la densité r, l'énergie E peut être soit positive (E > 0) soit négative (E< 0). В первом случае рассматриваемая галактика обладает достаточной кинетической энергией, чтобы преодолеть гравитационное притяжение массы М и удалиться на бесконечность. Это соответствует неограниченному монотонному расширению Вселенной (модель "открытой" Вселенной).
Dans le deuxième cas (E< 0) расширение Вселенной в какой-то момент прекратится и сменится сжатием (модель "замкнутой" Вселенной). Критическое значение плотности соответствует условию Е = 0, так что из (1.1) получаем:
rк = 3Н2 / 8pG (1,2).
En substituant dans cette expression les valeurs connues H = 15 ((km/s)/106 années-lumière) et G = 6,67×10-11 m3/kg s2, on obtient la valeur de la densité critique rc ~ 10-29 g /cm3. Ainsi, si l’Univers n’était constitué que de matière « visible » ordinaire avec une densité rв ~ 3 H 10-31 g/cm3, alors son avenir serait associé à une expansion illimitée. Cependant, comme mentionné ci-dessus, la présence de matière « noire » avec une densité rт > rв peut conduire à une évolution pulsée de l'Univers, lorsque la période d'expansion est remplacée par une période de compression (effondrement) (Fig. 1.2). Certes, récemment, les scientifiques arrivent de plus en plus à la conclusion que la densité de toute la matière dans l'Univers, y compris l'énergie « sombre », est exactement égale à la densité critique. Pourquoi est-ce ainsi ? Il n’y a pas encore de réponse à cette question.

Figure 1.2.
Le concept du Big Bang repose sur l’hypothèse que le début de l’évolution de l’Univers (t = 0) correspondait à un état de densité infinie r = Ґ (état singulier de l’Univers)1. A partir de ce moment, l'Univers s'étend2, et sa densité moyenne r diminue avec le temps selon la loi :
r~1/Gt2 (1.3)
où G est la constante gravitationnelle 3.
Le deuxième postulat de la théorie du Big Bang est la reconnaissance du rôle décisif du rayonnement lumineux sur les processus survenus au début de l’expansion4. La densité d'énergie e d'un tel rayonnement, d'une part, est liée à la température T par la formule bien connue de Stefan-Boltzmann :
où s = 7,6 10-16 J/m3 deg4 est la constante de Stefan-Boltzmann, et d'autre part, de densité de masse r :
r = e / с2 = sТ4/с2 (1,5)
où c est la vitesse de la lumière.
En substituant (1.6) dans (1.4), en tenant compte des valeurs numériques de G et s, on obtient :
T ~ 1010 t-1/2 (1,6)
où le temps est exprimé en secondes et la température en kelvins.
A très haute température (T > 1013 K, t< 10-6 с) Вселенная была абсолютно непохожа на то, что мы видим сегодня. В той Вселенной не было ни галактик, ни звезд, ни атомов... Как в "кипящем котле" в ней непрерывно рождались и исчезали кварки, лептоны и кванты фундаментальных взаимодействий, в первую очередь, фотоны (g). При столкновении двух фотонов могла, например, родиться пара электрон (е-) - позитрон (е+), которая практически сразу аннигилировала (самоуничтожалась), вновь рождая кванты света:
g + g " e- + e+ (1.7)
L'annihilation d'une paire électron-positon pourrait conduire à la naissance d'autres paires particule-antiparticule, par exemple neutrino (n) et antineutrino (n).
e- + e+ " n + `n (1.8)
Des réactions réversibles similaires se sont également produites avec la participation de hadrons, en particulier de nucléons (protons, neutrons et leurs antiparticules).
Il convient cependant de garder à l'esprit que la naissance d'une paire particule-antiparticule lors d'une collision de photons n'est possible qu'à la condition que l'énergie des photons Wg dépasse l'énergie restante W0 = m0c2 des particules en train de naître. L'énergie moyenne des photons en état d'équilibre thermodynamique est déterminée par la température :
où k est la constante de Boltzmann.
Par conséquent, le caractère réversible des processus impliquant des photons n'a eu lieu qu'à des températures dépassant une valeur bien spécifique pour chaque type de particules élémentaires T ~ m0c2/k.
Par exemple, pour les nucléons m0c2 ~ 1010 eV, ce qui signifie Tnucléon ~ 1013 K. Ainsi, à T > Tnucléon, l'apparition continue de paires nucléon-antinucléon et leur annihilation presque instantanée avec la naissance de photons pourraient se produire et se sont produites. Mais dès que la température T est devenue inférieure à T nucléon, les nucléons et antinucléons ont disparu en très peu de temps, se transformant en lumière. Et si tel était le cas pour tous les nucléons et antinucléons, alors l'Univers se retrouverait sans hadrons stables, et donc il n'y aurait aucune question à partir de laquelle les galaxies, étoiles et autres objets cosmiques se seraient ensuite formés. Mais il s’avère qu’en moyenne, pour chaque milliard de paires nucléon-antinucléon, il y avait une (!) particule « supplémentaire ». C’est à partir de ces nucléons « supplémentaires » que se construit la matière de notre Univers.
Un processus similaire d'annihilation des électrons et des positrons s'est produit plus tard, à t ~ 1 s, lorsque la température de l'Univers est tombée à ~ 1010 K et que l'énergie des photons est devenue insuffisante pour la création de paires électron-positon. En conséquence, un nombre relativement faible d’électrons est resté dans l’Univers – juste assez pour compenser la charge électrique positive des protons « supplémentaires ».
Les protons et les neutrons restant après l'autodestruction globale se sont convertis de manière réversible pendant un certain temps conformément aux formules de réaction :
p + e- " n + `n ;
p + n " n + e+ .
Et ici, le rôle décisif a été joué par la légère différence dans les masses au repos des protons et des neutrons, ce qui a finalement conduit au fait que les concentrations de neutrons et de protons se sont révélées différentes. La théorie affirme qu’à la fin de la cinquième minute, il y avait environ 15 neutrons pour cent protons. C'est à cette époque que la température de l'Univers est tombée à ~ 1010 K et que les conditions ont été créées pour la formation de noyaux stables, principalement des noyaux d'hydrogène (H) et d'hélium (He). Si nous négligeons les noyaux d'autres éléments (et ils ne sont presque pas apparus à l'époque), alors en tenant compte du rapport ci-dessus entre protons et neutrons, environ 70 % des noyaux d'hydrogène et environ 30 % des noyaux d'hélium auraient dû se former dans le Univers. C’est précisément ce rapport de ces éléments qui est observé dans le milieu intergalactique et dans les étoiles de première génération, confirmant ainsi le concept du Big Bang.
Après la formation des noyaux H et He, presque rien de remarquable ne s'est produit dans l'Univers pendant une longue période (de l'ordre d'un million d'années). Il faisait encore assez chaud pour que les noyaux retiennent les électrons, puisque les photons les arracheraient immédiatement. Par conséquent, l’état de l’Univers pendant cette période est appelé plasma de photons.
Cela a continué jusqu'à ce que la température chute à environ 4 000 K, et cela s'est produit environ 1 013 secondes, soit près d'un million d'années après le Big Bang. A cette température, les noyaux d'hydrogène et d'hélium commencent à capturer intensément les électrons et se transforment en atomes neutres stables (l'énergie des photons n'est plus suffisante pour briser ces atomes). Les astrophysiciens appellent ce processus la recombinaison.
Ce n'est qu'à partir de ce moment que la matière de l'Univers devient transparente au rayonnement et propice à la formation d'amas, à partir desquels se formeront plus tard des galaxies. Ce rayonnement, appelé rayonnement relique, mène depuis une existence indépendante, voyageant à travers l’Univers dans toutes les directions. Maintenant, des quanta de ce rayonnement nous parviennent sur Terre, qui ont parcouru presque en ligne droite une distance énorme égale au produit de la vitesse de la lumière c par le temps tр qui s'est écoulé depuis le moment de la recombinaison : L = сtр. Mais à la suite de l’expansion de l’Univers, nous « fuyons » en fait ces quanta de rayonnement de fond cosmique micro-onde à une vitesse v = НL ~ сtр/t0, où t0 = 1/Н est le temps qui s’est écoulé depuis l’apparition de l’Univers. Big Bang. Cela signifie que les longueurs d'onde du rayonnement de fond cosmique micro-ondes que nous recevons, en raison de l'effet Doppler, devraient être plusieurs fois (~ t0/tр) supérieures à celles qui étaient au moment de la recombinaison à T ~ 4000 K. Les calculs montrent que le rayonnement relique le rayonnement enregistré sur Terre devrait être le même que s'il était émis par un corps chauffé à une température T ~ 3 K1. Le rayonnement enregistré en 1965 par A. Penzias et R. Wilson possédait précisément ces propriétés.
PRÉVISION DU VOLUME CRITIQUE
où v sont des contributions partielles dont les valeurs, exprimées en cm cube 3 /mol, sont données dans le tableau. 5.2. Le calcul est assez simple et ne nécessite aucun commentaire supplémentaire.
PRÉDICTION DU FACTEUR ACENTRIQUE
Facteur acentrique
a été proposé en 1955 par Pitzer comme paramètre de corrélation caractérisant l'acentricité, ou non-sphéricité, d'une molécule. En analysant la dépendance de la pression réduite de vapeur saturée de diverses substances sur la température réduite, Pitzer et ses collègues ont découvert que pour l'argon, le krypton, le xénon, l'azote, l'oxygène, le monoxyde de carbone, le méthane et certaines autres substances, cette dépendance est décrite par presque une équation. Cependant, l'élargissement de cette liste avec des composés d'autres classes produit une série de lignes presque droites dont les pentes varient. Pitzer et al. ont adopté la pression de vapeur réduite. ![]() à une certaine température réduite
à une certaine température réduite ![]() comme caractéristique d'une substance. A ces températures, la pression réduite des gaz rares choisis comme substance simple est d'environ 0,1. Sur la base de cette observation, une définition d'un nouveau paramètre a été formulée : le facteur acentrique
comme décrivant l'écart de la valeur de la pression de vapeur réduite pour une certaine substance par rapport à la pression de vapeur réduite de la substance de comparaison sous la forme suivante :
comme caractéristique d'une substance. A ces températures, la pression réduite des gaz rares choisis comme substance simple est d'environ 0,1. Sur la base de cette observation, une définition d'un nouveau paramètre a été formulée : le facteur acentrique
comme décrivant l'écart de la valeur de la pression de vapeur réduite pour une certaine substance par rapport à la pression de vapeur réduite de la substance de comparaison sous la forme suivante :
(à T r =0,7),(5.18)
où est la pression de vapeur saturée de la substance à la température donnée T r =0,7.
Selon la définition de Pitzer, le facteur acentrique est « une mesure de l’écart des fonctions du potentiel intermoléculaire par rapport aux fonctions du potentiel intermoléculaire des molécules sphériques de la substance de référence ». Signification = 0 correspond à une symétrie sphérique dans un gaz raréfié. Les écarts par rapport au comportement caractéristique d'une substance simple sont évidents si > 0. Pour les gaz monoatomiques, le facteur acentrique est proche de zéro. Pour le méthane, c’est encore très faible. Cependant, pour les hydrocarbures de poids moléculaire élevé, la valeur augmente et augmente fortement avec l'augmentation de la polarité des molécules.
La plage de variation du facteur acentrique va de zéro à un. Actuellement, le facteur acentrique est largement utilisé comme paramètre qui caractérise dans une certaine mesure la complexité de la structure d'une molécule en ce qui concerne à la fois sa géométrie et sa polarité. Il est recommandé que l'applicabilité des corrélations incluant un facteur d'acentricité soit limitée aux gaz et liquides normaux et ne soit pas utilisée pour prédire les propriétés de liquides hautement polaires ou associés.
Il convient de noter ici que l'expérience de nos travaux nous permet de conclure que la limitation ci-dessus est trop catégorique. Sous réserve de certaines conditions de corrélation avec peut également être utilisé en relation avec les groupes nommés de substances organiques.
Les valeurs du facteur acentrique pour de nombreuses substances sont calculées sur la base des meilleures données expérimentales sur les pressions de vapeur, T c Et P. c connexions et sont contenus dans l’Annexe.
En l'absence d'informations sur pour le prédire, on peut utiliser :
Équation d'Edmister
 ;(5.19)
;(5.19)
équation de Lee-Kesler
Équation d'Ambrose-Walton
 ,(5.21)
,(5.21)
Où - pression critique, exprimé dans des atmosphères physiques;
= - point d'ébullition normal réduit de la substance ;
Le point d'ébullition normal d'une substance en degrés Kelvin ;
Température critique en degrés Kelvin.
f (0) , f (1) – défini dans la description de la méthode Ambrose-Walton (section 7.3)
Pour conclure notre examen du matériau sur les propriétés critiques et les critères de similarité, attardons-nous sur une autre question importante et générale. Il s'agit de critères de similarité. Actuellement, il y en a beaucoup qui sont proposés, nous avons fait connaissance avec l'un d'eux - le facteur acentrique. Insecte. 7 considère un autre critère de similarité - le coefficient de Riedel. Les deux critères sont très largement utilisés. Néanmoins, des approches universelles pour la sélection de l'un ou l'autre critère de similarité n'ont pas encore été créées, ce qui signifie que les travaux dans ce sens se poursuivront. Nous jugeons approprié de répéter les exigences énumérées par Wales dans sa monographie et liées à des paramètres supplémentaires ou à des critères de similarité :
Ces paramètres doivent être liés à la structure moléculaire et aux propriétés électrostatiques de la molécule.
Ils peuvent être déterminés avec un minimum de données expérimentales.
Les propriétés critiques ne doivent pas affecter directement leurs valeurs.
Lors de l'estimation de ces paramètres, il convient d'éviter d'utiliser des données sur PVT, sinon le sens de l'équation donnée est perdu.
Les paramètres supplémentaires doivent être fonction de la température, de préférence donnée.
Vous pouvez être d'accord ou en désaccord avec les exigences énumérées, mais il est bien évident que ni le facteur acentrique ni le critère de Riedel ne répondent à l'ensemble de leur complexe. De plus, il nous semble clair que l’une des raisons du succès de leur application est précisément la cohérence de leurs valeurs avec les paramètres critiques et les données P-T. Le support de la connexion avec les données P-T est la température d'ébullition à l'une des pressions, le plus souvent à la pression atmosphérique.
Ainsi, le développement de méthodes de prévision nécessitera probablement une clarification des exigences en matière de critères de similarité.
6. PRÉDICTION de la densité des gaz et des liquides
Avant de passer à la prédiction, il convient de rappeler que, selon la température et la pression adoptées, une substance peut être soit dans un état saturé, soit insaturé. La pression au-dessus d'un liquide saturé est égale à sa pression de vapeur saturée à une température donnée. La pression au-dessus d'un liquide insaturé, surfondu ou comprimé est supérieure à sa pression de vapeur saturée à la température choisie pour le calcul. Pour chacune des zones nommées PVT l’espace, il existe des approches indépendantes pour prédire la densité.
Prédire la densité de substances individuelles à l'aide du coefficient de compressibilité
Exemple 6.1
Pour l'isobutylbenzène, qui a une température critique de 650 K, une pression critique de 31 atm et un facteur acentrique de 0,378, calculez à l'aide des tableaux de Lee-Kesler (tableaux 4.6, 4.7) :
coefficient de compressibilité à 500, 657 et 1170 K et pression 1-300 atm,
densité à 500, 657 et 1170 K et pression 1-300 atm ;
donner des dépendances graphiques :
coefficient de compressibilité en fonction de la pression à des températures spécifiées,
densité en fonction de la pression à des températures spécifiées.
Solution
Nous utilisons le développement de Pitzer (équation 4.34) et le tableau. 4,6, 4,7 pour le coefficient de compressibilité.
Calculons les valeurs des températures données :
500/600 =0,769; = 657/650 =1,01; = 1170/650 =1,80.
Calculons les valeurs des pressions données :
1/31 =0,03226; = 300/31 =9,677.
Étant donné que la plage de pressions réduites d'intérêt coïncide avec la plage considérée par Lee-Kesler, nous utilisons des informations sur et pour les valeurs discrètes présentées dans le tableau. 4.6, 4.7.
Chacune des valeurs et est obtenue par interpolation linéaire par rapport à la température. Donc, à 500 K (= 0,769) et = 0,010 car on a
(0,9935-0,9922)/(0,80-0,75)·(0,769-0,75)+0,9922 = 0,9927.
Prédiction des densités de liquides saturés et de vapeur à l'aide d'équations d'étatsubstance
Trouver des conditions de saturation à partir des équations d'état est une tâche assez complexe, dont la solution est souvent impossible sans l'intervention de technologies informatiques et de logiciels spéciaux. Pour les équations d’état simples, comme l’équation de van der Waals, ce problème peut être résolu par des calculs simples. Cependant, il faut rappeler qu'en pratique, à l'aide de l'équation de Van der Waals, on ne peut évaluer que qualitativement l'état de saturation. Pour représenter plus précisément la saturation, d'autres équations d'état et méthodes spéciales ont été développées.
Dans ce manuel, en utilisant l'équation de van der Waals comme exemple, nous envisageons une approche pour trouver la pression de saturation et les volumes de saturation de liquide et de vapeur (points appartenant au binodal), ainsi que les conditions qui déterminent les états métastables de la matière. (points extrêmes isothermes).
AGENCE FÉDÉRALE POUR L'ÉDUCATION
SUCCURSALE D'UN ÉTABLISSEMENT ÉDUCATIF D'ÉTAT
FORMATION PROFESSIONNELLE SUPÉRIEURE
"PÉTROLE D'ÉTAT D'UFA
UNIVERSITÉ TECHNIQUE" à Salavat
Branche de l'établissement d'enseignement public d'enseignement professionnel supérieur USPTU à Salavat
Département des procédés technologiques chimiques
CALCUL DES PHYSIQUES CRITIQUES ET THERMIQUES
PROPRIÉTÉS ET MASSE MOLÉCULAIRE DES SUBSTANCES
Manuel pédagogique et méthodologique
Le manuel pédagogique et méthodologique est destiné aux étudiants des établissements d'enseignement supérieur des spécialités d'ingénierie qui étudient le cours « Calculs techniques des propriétés physiques et chimiques des substances ».
Le manuel présente quelques méthodes de calcul des propriétés thermophysiques critiques et du poids moléculaire des substances, ainsi que des exemples de calcul.
Compilé par : Sidracheva I. I., assistant
Assistant
Introduction
Lors des calculs technologiques des processus et appareils de raffinage du pétrole, une connaissance des propriétés physiques et chimiques des substances chimiques et des fractions pétrolières est requise.
La littérature de référence fournit généralement des données sur les propriétés physicochimiques des substances pour un nombre limité de composés les plus courants. À cet égard, se pose le problème du calcul analytique des valeurs numériques des propriétés physico-chimiques des substances et de la prise en compte de l'influence des paramètres de procédé sur les propriétés des substances.
Ce manuel pédagogique propose un certain nombre de méthodes pour calculer les propriétés physiques et chimiques de base des composés chimiques et des fractions pétrolières. Avec son aide, les étudiants peuvent maîtriser la méthodologie de calcul et comparer diverses méthodes pour déterminer avec précision les propriétés des substances.
1 CALCUL DES PARAMÈTRES CRITIQUES DES SUBSTANCES
Une partie importante des méthodes d'ingénierie permettant de calculer les propriétés physiques et chimiques des substances ne repose pas sur les valeurs réelles des paramètres, mais sur leurs valeurs réduites. Les paramètres donnés désignent le rapport entre la valeur réelle du paramètre et sa valeur critique. Par exemple, pour une température donnée :
où est la température critique pour une substance donnée.
À cet égard, une détermination assez fiable des paramètres critiques des substances est nécessaire pour un calcul plus correct des propriétés physicochimiques des substances.
L'état d'une substance dans lequel la différence entre ses phases liquide et gazeuse disparaît est dit critique.
La température critique est la température maximale à laquelle les phases vapeur et liquide peuvent encore coexister en équilibre. À des températures supérieures au point critique, la condensation des vapeurs de cette substance est impossible. Au point critique, les valeurs de pression critique et de volume spécifique critique sont fixées.
Les paramètres critiques d'une substance sont liés par la relation
où est le coefficient de compressibilité critique ;
constante universelle des gaz.
Selon les données expérimentales, le coefficient de compressibilité critique varie entre 0,26 et 0,29 (pour la plupart des composés organiques), bien qu'il existe des exceptions.
La température critique, la pression et le volume sont trois constantes largement utilisées pour les substances pures. Cependant, leurs mesures modernes ne sont presque jamais effectuées.
Considérons les méthodes de calcul pour déterminer les propriétés critiques des composants purs.
1.1 CALCUL DE LA TEMPÉRATURE CRITIQUE
La température critique d'une substance pure est la température maximale à laquelle les phases liquide et vapeur peuvent exister en équilibre.
La température critique d'un liquide Tcr (K) peut être déterminée approximativement en connaissant le point d'ébullition des liquides Tcr (K) à pression atmosphérique.
1.1. La règle de Guldberg :
1.2. Règle de Guldberg modifiée :
1.3. Méthode Meissner – Redding : pour les connexions avec Tk<235 К и простых веществ:
![]()
Pour les connexions avec Tc compris entre 235 et 600 K, vous pouvez utiliser les équations :
1.3.1. Pour les alcanes et alcènes :
1.3.2. Pour les hydrocarbures aromatiques et les naphtènes sans halogènes ni soufre :
où est le rapport entre le nombre d’atomes de carbone situés à l’extérieur du cycle et le nombre total d’atomes de carbone dans le composé.
1.3.3. Pour les composés contenant des atomes d’halogène et de soufre :
où est le nombre d’atomes d’halogène ou de soufre dans la molécule.
1.4. Pour les hydrocarbures, vous pouvez utiliser l'équation de Goethe et Thodos :
![]() (1.7)
(1.7)
1.5. Pour les hydrocarbures n-alcanes, la température critique est calculée à l'aide de l'équation de Tilicheev et Tatevsky :
 (1.8)
(1.8)
où est le nombre d'atomes de carbone.
1.6. Vous pouvez utiliser les équations de Mamedov :
1.6.1. Pour les hydrocarbures n-alcanes :
![]() (1.9)
(1.9)
où est le poids moléculaire de l’hydrocarbure.
1.6.2. Pour les hydrocarbures aromatiques - homologues du benzène :
 (1.10)
(1.10)
où est le point d'ébullition molaire moyen de la fraction liquide lors de la distillation ;
densité d'hydrocarbure liquide à 288 K.
1.7. La température critique des hydrocarbures paraffiniques normaux est déterminée par l'équation
1.8. La température critique peut être déterminée par la formule de Nokai
où est la densité du liquide, kg/m3.
1.9. Méthode Lidersen :
Les valeurs sont tirées du tableau 1.1.
1.10. La température critique des hydrocarbures et des fractions pétrolières est également calculée à l'aide de l'équation d'Eaton et Porter :
où T50% est le point d'ébullition de 50% de la fraction selon ASTM.
Tableau 1.1 – Composants permettant de déterminer les paramètres critiques
(selon Leaderson)
|
Atome, groupe, connexion |
|
||
|
CH3 et -CH2- | |||
|
CH2- (dans le ring) | |||
|
CH (sur le ring) | |||
|
C - (sur le ring) | |||
|
CH (sur le ring) | |||
|
C-(sur le ring) | |||
|
C= (sur le ring) | |||
|
OH (alcools) | |||
|
OH (phénols) | |||
|
O - (sur le ring) | |||
Suite du tableau 1.1
Note. Les données peu fiables sont indiquées entre parenthèses.
1.11. D'après l'équation de Maxwell :
où et sont des coefficients empiriques.
1.12. La température critique des liquides peut être déterminée par la dépendance
 , (1.18)
, (1.18)
où est la densité du liquide aux températures T1 et T2, g/cm3.
La portée de l'équation (1.18) est limitée par les conditions suivantes :
![]() ;
;
![]()
De plus, plus l’intervalle de température T est petit, plus l’écart par rapport aux données expérimentales est grand.
1.13. La méthode de Filippov :
où est la densité critique, g/cm3.
 (1.20)
(1.20)
où est la densité à température, g/cm3.
1.2 CALCUL DE LA PRESSION CRITIQUE
La pression critique est la pression à laquelle une substance peut encore être à l'état liquide à une température critique, c'est-à-dire la pression de vapeur saturée à une température critique.
1.2.1. La pression critique des liquides peut être calculée à l'aide de la formule de Riedel :
où Pcr est la pression critique, Pa.
Les composants permettant de déterminer a sont tirés du tableau 1.2. Ces composantes se résument avec une constante A=105.4∙10-3.
Tableau 1.2 - Composantes pour déterminer a (selon la méthode de Riedel)
1.2.2. D'après la formule de Lydersen – Riedel :
 (1.22)
(1.22)
où ΣΔр est déterminé en additionnant les composants donnés dans le tableau 1.1.
2.3. D'après la formule de Behnke :
![]() (1.23)
(1.23)
où est le nombre d'atomes dans la molécule.
2.4. Dépendance au parachor Pch et à la réfraction molaire Rd selon Meissner :
 (1.24)
(1.24)
La valeur de Pch et Rd est déterminée en additionnant les composants donnés dans les tableaux 1.3 et 1.4.
Tableau 1.3 - Composants pour déterminer le parachor
|
Atome, groupe ou lien |
Composant ∙ 103,
|
|
CH2 à (CH2)n : | |
|
O (à l'antenne) | |
|
Double liaison : bout de chaîne 2 ou 3 positions 3, 4 positions | |
|
Triple liaison | |
|
trois mandats quatre mandats à cinq membres six membres |
Tableau 1.4 - Composants permettant de déterminer la réfraction molaire
2.5. La pression critique en fonction de la température critique peut être calculée à l'aide de l'équation de Lewis :
![]() (1.25)
(1.25)
où K est une constante, tirée du tableau 1.5 ou pour les fractions pétrolières calculée à l'aide de la formule
![]() (1.26)
(1.26)
où T10%, T70% sont les températures d'ébullition des fractions 10 et 70%, K.
2.6. Pression critique selon la formule de Filippov :
![]() (1.27)
(1.27)
où ρcr est la densité critique de la substance, en kg/m3, définie comme
 (1.28)
(1.28)
où ρt, ρ1, ρ2 sont les densités de la substance aux températures T, T1, T2, g/cm3.
Tableau 1.5 – Valeurs de la constante K dans l'équation de Lewis
Ou la densité molaire critique d'une substance est calculée à l'aide de la formule
![]() (1.29)
(1.29)
où R est la constante universelle des gaz égale à 8,314∙103 J/(kmol∙K).
1.3 CALCUL DU VOLUME CRITIQUE
Le volume occupé par une substance à une pression et une température critiques est dit critique.
Le volume critique ne peut pas être déterminé avec précision car au point critique, des changements de pression négligeables entraînent des changements de volume importants.
3.1. Le volume critique selon la méthode Leaderson est déterminé par la formule
![]() (1.30)
(1.30)
où Vcr est le volume critique, m3/kmol.
Les valeurs ΔV sont tirées du tableau 1.1.
3.2. Méthode de Vetere, similaire à la méthode Leaderson :
 (1.31)
(1.31)
où les valeurs ΔVi pour de nombreux groupes sont données dans le tableau 1.6,
Mi est le poids moléculaire du groupe,
Vcr – volume critique, cm3/mol.
3.3. Le volume critique peut être déterminé à partir du coefficient de compressibilité critique Zcr :
![]() , (1.32)
, (1.32)
![]() . (1.33)
. (1.33)
Les valeurs Δz proviennent du tableau 1.7.
3.4. Le volume critique d'une substance en fonction du parachor peut être calculé à l'aide de la formule suivante :
3.5. Selon Meissner :
3.6. La formule Todos est utilisée pour déterminer Vcr des hydrocarbures aliphatiques saturés et insaturés :
![]() (1.36)
(1.36)
où in est la constante de van der Waals, déterminée par l'équation
![]() (1.37)
(1.37)
où B – dépend du nombre d'atomes de carbone (Nc) et de leur emplacement dans la molécule :
pour les hydrocarbures aliphatiques saturés normaux :
B=0,7849 – 0,01337·Nc ; (1,38)
pour les mêmes hydrocarbures, mais à chaînes ramifiées :
B=0,8100 – 0,0138·Nc ; (1,39)
pour les hydrocarbures aliphatiques insaturés :
![]() (1.40)
(1.40)
Tableau 1.6 - Composants de groupe pour la détermination du volume critique par la méthode Vetere
|
Acyclique (en dehors du ring) |
C=O (dans le ring) | ||
|
Dans la chaîne principale : |
|||
|
CH3, CH2, CH, C |
HC=O (aldéhydes) | ||
|
Dans la chaîne latérale : |
|||
|
CH3, CH2, CH, C |
NH (hors du ring) | ||
|
NH (en anneau) | |||
|
N - (en dehors du ring) | |||
|
Cyclique (en anneau) |
|||
|
N - (sur le ring) | |||
|
S - (en dehors du ring) | |||
|
S - (sur le ring) | |||
|
OH (alcools) | |||
|
OH (phénols) |
C=O (en dehors du ring) | ||
|
O-(en dehors du ring) | |||
|
O - (sur le ring) | |||
|
O-(époxydes) |
Tableau 1.7 – Composantes de Z
|
Groupe ou connexion |
Groupe ou connexion | ||
|
CH3 ou - CH2 | |||
|
Les 10 premiers de la molécule |
Liaison C=C | ||
|
Tous les dix | |||
|
CH ou -C- : |
alicyclique aromatique | ||
|
D'abord dans la molécule |
NH (aliphatique) | ||
|
Chaque suivant |
N - (aliphatique) | ||
|
CH (sur le ring) | |||
|
trois mandats | |||
|
Cinq et six membres |
O - (sur le ring) | ||
|
Anneau benzénique |
HCOO - (esters de l'acide formique) : | ||
|
Dispositions |
À 4 ou moins | ||
|
COO-(esters) à< 5 атомах à > 5 atomes | |||
|
Relation C=C : | |||
|
(aromatique) |
1.4 EXEMPLES DE CALCUL
Exemple 1.4.1. Calculer la température critique de l'éther éthylbutylique à l'aide de la méthode Leaderson, si Tk = 365,4 K.
En utilisant les données du tableau 1.1 de l'annexe, nous trouvons
D'après l'équation (1.14) :
D'après l'équation (1.15) :
![]()
D'après les données de la littérature, Tcr = 531 K.
Exemple 1.4.2. Calculez la température critique de l'éthylbenzène à l'aide de la méthode Nokai, si Tk = 409,3 K, ρ = 0,867 g/cm3.
D'après l'équation (1.12) :
Tcr = 615,2 K.
D'après les données de la littérature, Tcr = 617,1 K.
Exemple 1.4.3. Calculez la température critique à l'aide de la méthode Meissner – Redding pour l'isobutylbenzène si Tk = 445,9 K.
D'après l'équation (1.5) :
Tcr = 1,41∙445,9 + 66 – 0,4∙(0,383∙445,9 – 93) = 663,6 (K).
D'après les données de la littérature, Tcr = 650 K.
Exemple 1.4.4. Calculez Pcr et Vcr du trichloroéthane si Tcr = 602 K.
Rcr est déterminé à l'aide de la formule de Meissner. Résumons les composants de Pch selon le tableau 2.2 de l'annexe.
(J1/4m5/2kmol-1).
Résumons les composantes de Rd selon le tableau 2.3 de l'annexe.
(m3/kmol).
En utilisant l’équation (2.4), nous calculons Pcr :
D'après les données de la littérature Pcr = 4,1 MPa.
En utilisant la méthode parachor (3.5), nous déterminons Vcr :
(m3/kmol).
D'après les données de la littérature, Vcr = 0,294 m3/kmol.
Exemple 1.4.5. Déterminez Tcr, Pcr de l'heptane, si M = 100 kg/kmol, à 273 K, ρ0 = 0,7005 g/cm3 et à 293 K ρ = 0,6836 g/cm3.
En utilisant l’équation (1.20), nous déterminons ρcr :
D'après l'équation (1.19) :
En utilisant l’équation (2.7), nous déterminons Pcr :
D'après les données de la littérature Tcr = 540,2 K, Pcr = 2,7 MPa.
Exemple 1.4.6. Calculer Vcr de l'éther éthylpropylique ; Tcr = 498 K, Pcr = 3,23 MPa.
À l’aide de l’équation de Leaderson (3.1) et du tableau annexe 1.1 :
M3/kmol.
M3/kmol.
En revanche, d'après l'équation (3.4) et le tableau 3.2 de l'annexe :

Alors selon l’équation (3.3) :
M3/kmol.
La valeur expérimentale est de 0,339 m3/kmol.
2 CALCUL DE LA MASSE MOLÉCULAIRE DES FRACTIONS D'HUILE
Le poids moléculaire est l’une des principales caractéristiques physiques et chimiques des huiles et des produits pétroliers. Il est utilisé pour calculer d'autres propriétés physiques et chimiques, ainsi que pour les calculs technologiques des équipements.
Le poids moléculaire des substances individuelles est facilement calculé à partir de leur formule chimique et des masses atomiques des éléments qui composent les molécules.
Le poids moléculaire moyen d'un mélange peut être déterminé en connaissant la fraction molaire et le poids moléculaire de chaque composant du mélange :
où est la teneur en composants du mélange, en fractions molaires ;
![]() poids moléculaires des composants du mélange.
poids moléculaires des composants du mélange.
Si ce ne sont pas les fractions molaires, mais les masses des composants du mélange qui sont connues, vous pouvez utiliser la formule suivante :
 , (2.2)
, (2.2)
Où ![]() masse des composants, kg ;
masse des composants, kg ;
![]() poids moléculaire des composants.
poids moléculaire des composants.
En ce qui concerne les systèmes pétroliers qui sont des mélanges à plusieurs composants, le poids moléculaire signifie le poids moléculaire moyen - le poids moléculaire d'un hydrocarbure hypothétique ayant des valeurs moyennes de composition élémentaire, de point d'ébullition et de densité.
Il existe une certaine relation entre le poids moléculaire et le point d'ébullition des fractions pétrolières : plus le poids moléculaire de la fraction pétrolière est élevé, plus son point d'ébullition est élevé. Parmi les méthodes de calcul permettant de déterminer le poids moléculaire, la formule la plus largement utilisée est une formule raffinée basée sur cette dépendance.
où est la température d'ébullition moléculaire moyenne (pour les fractions étroites, vous pouvez prendre la température d'ébullition moyenne pendant la distillation selon GOST), ˚С ;
facteur caractérisant.
Le facteur caractérisant détermine la nature chimique du produit pétrolier. Pour les produits pétroliers paraffiniques K = 12,5 – 13, pour les produits pétroliers aromatisés environ 10 ou moins, pour les produits pétroliers naphténiques-aromatiques K = 10 – 11. Le facteur caractéristique est déterminé par la formule :
 , (2.4)
, (2.4)
où est la densité relative de cette fraction.
Le point d'ébullition moléculaire moyen d'un produit pétrolier peut être déterminé par la formule :
![]() , (2.5)
, (2.5)
où est la teneur en fractions étroites, fractions molaires ;
températures moyennes (arithmétiques) de début et de fin d'ébullition des fractions étroites qui composent ce produit pétrolier, ˚С.
Si seule la densité du produit pétrolier est connue, son poids moléculaire peut être déterminé à l'aide de la formule de Craig :
![]() . (2.6)
. (2.6)
3 CALCUL DES PROPRIÉTÉS THERMIQUES PHYSIQUES DES SUBSTANCES
Les calculs technologiques thermiques des équipements des raffineries de pétrole reposent sur des caractéristiques thermophysiques de substances telles que la capacité thermique, la chaleur de vaporisation et de condensation, l'enthalpie (teneur calorifique), la chaleur de combustion, etc.
3.1 CALCUL DE LA CHALEUR DE FORMATION DE VAPEUR
La chaleur de vaporisation ou la chaleur de condensation, de signe opposé, est la différence entre les enthalpies de la vapeur saturée et son liquide bouillant à l'équilibre.
Sur la base des équations d'état, un certain nombre d'équations simples mais assez précises ont été développées pour calculer la chaleur de vaporisation. Par exemple, l'équation de Chen
et l'équation de Riedel
![]() (3.2)
(3.2)
Exemple 3.1.1 Calculez la chaleur de vaporisation de l'hexane à un point d'ébullition normal de 341,9 K (1 atm) à l'aide des méthodes de Chen et Riedel. La température critique de l'hexane est de 507,3 K, la pression critique est de 29,9 atm. La valeur tabulée de la chaleur de vaporisation de l’hexane est de 6896 cal/mol.
Selon la méthode de Chen
Erreur de calcul.
Selon la méthode Riedel

Erreur de calcul
3.2 CALCUL DE LA CAPACITÉ THERMIQUE
La capacité thermique est la quantité de chaleur qui doit être transmise à une substance afin d'augmenter la température d'une unité quantitative de 1 °C.
Selon l'unité quantitative choisie pour la substance, on distingue la capacité thermique molaire (en kJ/(kmol K) ou kcal/(kmol K), la capacité thermique massique (en kJ/kg K ou kcal/kg K) ou capacité thermique volumétrique (en kJ /m3·K ou kcal/m3·K).
3.2.1 CALCUL DE LA CAPACITÉ THERMIQUE DES SUBSTANCES EN PHASE VAPEUR
Pour calculer théoriquement la capacité thermique, la connaissance de données structurelles et spectrales détaillées est nécessaire. La capacité thermique d’une substance fait référence à l’état d’un gaz parfait. Pour passer aux gaz réels, on utilise souvent le terme « pression nulle », dans laquelle les équations d'état des gaz parfaits et réels coïncident. Pour les calculs techniques de la capacité thermique, une équation thermique est généralement utilisée sous la forme d'une expansion de la dépendance de la capacité thermique à la température dans une série polynomiale
où est la capacité thermique spécifique à « pression nulle », cal/(mol deg) ;
T – température. À;
Constantes.
et pour des températures élevées
où est le nombre d’atomes de carbone et d’hydrogène dans la molécule.
3.2.2 calcul de la capacité thermique des substances en phase liquide
La plupart des liquides organiques ont une capacité thermique comprise entre 0,4 et 0,5 cal/(g K), la capacité thermique augmente légèrement avec l'augmentation de la température. L'effet de la pression sur la capacité thermique d'un liquide, à l'exception de la région critique, est également faible et pour la plupart des liquides à faible point d'ébullition, la capacité thermique diminue d'environ 10 % lorsque la pression augmente de 2 500 atm.
La méthode de groupe additif pour calculer la capacité thermique, développée par Johnson et Huang, est assez simple, dans laquelle les composants du groupe correspondant de la molécule (tableau 3.2) sont simplement résumés.
Tableau 3.2 - Composants du groupe atomique pour le calcul de la capacité thermique des liquides à 20 ˚С selon la méthode Johnson et Huang
Formule développée du 2-méthylpentane
H3C–CH–CH2–CH2–CH3
Nous présentons le calcul de la capacité thermique sous forme de tableau :
La capacité calorifique massique du 2-méthylpentane est :
![]() cal/(gK).
cal/(gK).
3.3 CALCUL DE L'ENTHALPIE
L'enthalpie caractérise la quantité d'énergie thermique par unité de masse d'une substance à une température spécifique et se mesure en J/g ou cal/g. La méthodologie de calcul de l'énergie des molécules est assez complexe et l'énergie des molécules n'est déterminée que pour les molécules les plus simples constituées de plusieurs atomes. À cet égard, dans les calculs techniques liés à la nécessité de déterminer la quantité de chaleur qui doit être introduite ou retirée d'un système technologique pour mettre en œuvre un processus technologique, une enthalpie conditionnelle égale à zéro à 0 ˚C est considérée comme la référence de température. indiquer.
À la température T, l'enthalpie d'une substance en phase liquide ou gazeuse est calculée comme le produit de la capacité thermique de la substance et de la température.
Les équations de calcul des enthalpies des hydrocarbures en kJ/kg dans les phases liquide et vapeur en fonction de la température t (˚С) et de la densité relative de la substance pour des pressions de 1 à 10 atmosphères se sont généralisées dans la pratique du calcul :
pour phase liquide
 , (3.5)
, (3.5)
pour la phase vapeur
RÉFÉRENCES
1. Calculs Viktorov des grandeurs physiques et chimiques et calculs appliqués - Leningrad : Chimie, 1977.-360 p.
2. , Rakhimov définit les propriétés critiques des composants purs. Lignes directrices pour les calculs techniques des propriétés physiques et chimiques des substances. – Oufa : USNTU, 1995. – 16 p.
3. , Ilyin, méthodes de calcul des propriétés physiques et chimiques des substances : Manuel. – Oufa : USNTU, 2004. – 176 p.
4. Reed R., Prausnitz J., Sherwood T. Propriétés des gaz et des liquides : Un manuel de référence / Trad. De l'anglais Éd. .- éd., révisé. et complémentaire - L. : Chimie, 1982.-592 p.
5. Calculs des principaux procédés et appareils de raffinage du pétrole : Manuel /, etc. : Ed. . -3e éd., révisée. et supplémentaire – M. : Chimie, 1979. –568 p.
6. , Propriétés physiques et chimiques d'Orlova des liquides : Manuel. – L. : Chimie, 1976. – 112 p.
Introduction 1
1. CALCUL DES PARAMÈTRES CRITIQUES DES SUBSTANCES 2
2. CALCUL DE LA MASSE MOLÉCULAIRE DES FRACTIONS D'HUILE 19
3. CALCUL DES PROPRIÉTÉS THERMIQUES PHYSIQUES DES SUBSTANCES 21
RÉFÉRENCES 28
L'univers est tout ce qui existe. Des plus petits grains de poussière et atomes aux énormes accumulations de matière des mondes et systèmes stellaires. Ce ne serait donc pas une erreur de dire que toute science, d'une manière ou d'une autre, étudie l'Univers, plus précisément l'un ou l'autre de ses aspects. Il existe une discipline scientifique dont l'objet d'étude est l'Univers lui-même. Il s’agit d’une branche particulière de l’astronomie, appelée cosmologie.
La cosmologie est l'étude de l'Univers dans son ensemble, qui comprend la théorie de l'ensemble de la région couverte par les observations astronomiques comme faisant partie de l'Univers.
Avec le développement de la science, qui révèle de plus en plus les processus physiques qui se produisent dans le monde qui nous entoure, la plupart des scientifiques se sont progressivement tournés vers des idées matérialistes sur l'infinité de l'Univers. Ici, la découverte par I. Newton (1643 - 1727) de la loi de la gravitation universelle, publiée en 1687, fut d'une grande importance. L'une des conséquences importantes de cette loi fut l'affirmation selon laquelle dans un Univers fini, toute sa matière doit être. être rassemblé en un seul système fermé dans une période de temps limitée, puis comment dans un Univers infini, la matière sous l'influence de la gravité est collectée dans certains volumes limités (selon les idées de l'époque - dans les étoiles), remplissant uniformément le Univers.
La théorie générale de la relativité, créée par A. Einstein (1879 - 1955), revêt une grande importance pour le développement des idées modernes sur la structure et le développement de l'Univers. Il généralise la théorie de la gravité de Newton aux grandes masses et aux vitesses comparables à la vitesse de la lumière. En effet, une masse colossale de matière est concentrée dans les galaxies, et les vitesses des galaxies lointaines et des quasars sont comparables à la vitesse de la lumière.
L'une des conséquences importantes de la théorie de la relativité générale est la conclusion sur le mouvement continu de la matière dans l'Univers - la non-stationnarité de l'Univers. Cette conclusion a été obtenue dans les années 20 de notre siècle par le mathématicien soviétique A.A. Friedman (1888 - 1925). Il a montré que, selon la densité moyenne de la matière, l'Univers devrait soit se dilater, soit se contracter. À mesure que l'Univers s'étend, la vitesse à laquelle les galaxies s'éloignent devrait être proportionnelle à la distance qui les sépare - une conclusion confirmée par Hubble par la découverte du décalage vers le rouge dans le spectre des galaxies.
La valeur critique de la densité moyenne d'une substance, dont dépend la nature de son mouvement,
où G est la constante gravitationnelle et H=75 km/s*Mpc est la constante de Hubble. En substituant les valeurs requises, nous constatons que la valeur critique de la densité moyenne de la substance est P k = 10 -29 g/cm 3 .
Si la densité moyenne de matière dans l'Univers est supérieure à la densité critique, alors à l'avenir l'expansion de l'Univers sera remplacée par une compression, et si la densité moyenne est égale ou inférieure à la densité critique, l'expansion ne sera pas arrêt. Une chose est claire : au fil du temps, l'expansion a entraîné une diminution significative de la densité de la matière, et à un certain stade d'expansion, des galaxies et des étoiles ont commencé à se former.