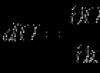There are four levels structural organization proteins: primary, secondary, tertiary and quaternary. Each level has its own characteristics.
The primary structure of proteins is a linear polypeptide chain of amino acids connected by peptide bonds. Primary structure is the simplest level of structural organization of a protein molecule. High stability is given to it by covalent peptide bonds between the α-amino group of one amino acid and the α-carboxyl group of another amino acid. [show] .
If the imino group of proline or hydroxyproline is involved in the formation of a peptide bond, then it has a different form [show] .

When peptide bonds form in cells, the carboxyl group of one amino acid is first activated, and then it combines with the amino group of another. Laboratory synthesis of polypeptides is carried out in approximately the same way.
A peptide bond is a repeating fragment of a polypeptide chain. It has a number of features that affect not only the shape of the primary structure, but also the higher levels of organization of the polypeptide chain:


- coplanarity - all atoms included in the peptide group are in the same plane;
- the ability to exist in two resonance forms (keto or enol form);
- trans position of the substituents relative to the C-N bond;
- the ability to form hydrogen bonds, and each of the peptide groups can form two hydrogen bonds with other groups, including peptide ones.
The exception is peptide groups involving the amino group of proline or hydroxyproline. They are only able to form one hydrogen bond (see above). This affects the formation of the secondary structure of the protein. The polypeptide chain in the area where proline or hydroxyproline is located easily bends, since it is not held, as usual, by a second hydrogen bond.
Nomenclature of peptides and polypeptides . The name of peptides is made up of the names of their constituent amino acids. Two amino acids make a dipeptide, three make a tripeptide, four make a tetrapeptide, etc. Each peptide or polypeptide chain of any length has an N-terminal amino acid containing a free amino group and a C-terminal amino acid containing a free carboxyl group. When naming polypeptides, all amino acids are listed sequentially, starting with the N-terminal one, replacing in their names, except for the C-terminal one, the suffix -in with -yl (since the amino acids in peptides no longer have a carboxyl group, but a carbonyl one). For example, the name shown in Fig. 1 tripeptide - leuc silt phenylalane silt threon in.
Features of the primary structure of the protein . In the backbone of the polypeptide chain, rigid structures (flat peptide groups) alternate with relatively mobile regions (-CHR), which are capable of rotating around bonds. Such structural features of the polypeptide chain affect its spatial arrangement.
Secondary structure is a way of folding a polypeptide chain into an ordered structure due to the formation of hydrogen bonds between peptide groups of the same chain or adjacent polypeptide chains. According to their configuration, secondary structures are divided into helical (α-helix) and layered-folded (β-structure and cross-β-form).


α-Helix. This is a type of secondary protein structure that looks like a regular helix, formed due to interpeptide hydrogen bonds within one polypeptide chain. The model of the structure of the α-helix (Fig. 2), which takes into account all the properties of the peptide bond, was proposed by Pauling and Corey. Main features of the α-helix:
- helical configuration of the polypeptide chain having helical symmetry;
- the formation of hydrogen bonds between the peptide groups of each first and fourth amino acid residue;
- regularity of spiral turns;
- the equivalence of all amino acid residues in the α-helix, regardless of the structure of their side radicals;
- side radicals of amino acids do not participate in the formation of the α-helix.
Externally, the α-helix looks like a slightly stretched spiral of an electric stove. The regularity of hydrogen bonds between the first and fourth peptide groups determines the regularity of the turns of the polypeptide chain. The height of one turn, or the pitch of the α-helix, is 0.54 nm; it includes 3.6 amino acid residues, i.e., each amino acid residue moves along the axis (the height of one amino acid residue) by 0.15 nm (0.54:3.6 = 0.15 nm), which allows us to talk about equivalence of all amino acid residues in the α-helix. The regularity period of an α-helix is 5 turns or 18 amino acid residues; the length of one period is 2.7 nm. Rice. 3. Pauling-Corey a-helix model
β-Structure. This is a type of secondary structure that has a slightly curved configuration of the polypeptide chain and is formed by interpeptide hydrogen bonds within individual areas one polypeptide chain or adjacent polypeptide chains. It is also called a layered-fold structure. There are varieties of β-structures. The limited layered regions formed by one polypeptide chain of a protein are called cross-β form (short β structure). Hydrogen bonds in the cross-β form are formed between the peptide groups of the loops of the polypeptide chain. Another type - the complete β-structure - is characteristic of the entire polypeptide chain, which has an elongated shape and is held by interpeptide hydrogen bonds between adjacent parallel polypeptide chains (Fig. 3). This structure resembles the bellows of an accordion. Moreover, variants of β-structures are possible: they can be formed by parallel chains (the N-terminal ends of the polypeptide chains are directed in the same direction) and antiparallel (the N-terminal ends are directed in the same direction) different sides). The side radicals of one layer are placed between the side radicals of another layer.
In proteins, transitions from α-structures to β-structures and back are possible due to the rearrangement of hydrogen bonds. Instead of regular interpeptide hydrogen bonds along the chain (thanks to which the polypeptide chain is twisted into a spiral), the helical sections unwind and hydrogen bonds close between the elongated fragments of the polypeptide chains. This transition is found in keratin, the protein of hair. When washing hair with alkaline detergents, the helical structure of β-keratin is easily destroyed and it turns into α-keratin (curly hair straightens).
The destruction of the regular secondary structures of proteins (α-helices and β-structures), by analogy with the melting of a crystal, is called the “melting” of polypeptides. In this case, hydrogen bonds are broken, and the polypeptide chains take the form of a random tangle. Consequently, the stability of secondary structures is determined by interpeptide hydrogen bonds. Other types of bonds take almost no part in this, with the exception of disulfide bonds along the polypeptide chain at the locations of cysteine residues. Short peptides are closed into cycles due to disulfide bonds. Many proteins contain both α-helical regions and β-structures. There are almost no natural proteins consisting of 100% α-helix (the exception is paramyosin, a muscle protein that is 96-100% α-helix), while synthetic polypeptides have 100% helix.
Other proteins have varying degrees of coiling. A high frequency of α-helical structures is observed in paramyosin, myoglobin, and hemoglobin. In contrast, in trypsin, a ribonuclease, a significant part of the polypeptide chain is folded into layered β-structures. Proteins of supporting tissues: keratin (protein of hair, wool), collagen (protein of tendons, skin), fibroin (protein of natural silk) have a β-configuration of polypeptide chains. Various degrees helicalization of polypeptide chains of proteins indicates that, obviously, there are forces that partially disrupt helicalization or “break” the regular folding of the polypeptide chain. The reason for this is a more compact folding of the protein polypeptide chain in a certain volume, i.e., into a tertiary structure.
Protein tertiary structure
The tertiary structure of a protein is the way the polypeptide chain is arranged in space. Based on the shape of their tertiary structure, proteins are mainly divided into globular and fibrillar. Globular proteins most often have an ellipsoid shape, and fibrillar (thread-like) proteins have an elongated shape (rod or spindle shape).
However, the configuration of the tertiary structure of proteins does not yet give reason to think that fibrillar proteins have only a β-structure, and globular proteins have an α-helical structure. There are fibrillar proteins that have a helical, rather than layered, folded secondary structure. For example, α-keratin and paramyosin (protein of the obturator muscle of mollusks), tropomyosins (proteins skeletal muscles) belong to fibrillar proteins (have a rod-shaped form), and their secondary structure is an α-helix; in contrast, globular proteins may contain a large number of β-structures.
Spiralization of a linear polypeptide chain reduces its size by approximately 4 times; and packing into the tertiary structure makes it tens of times more compact than the original chain.
Bonds that stabilize the tertiary structure of a protein . Bonds between side radicals of amino acids play a role in stabilizing the tertiary structure. These connections can be divided into:
- strong (covalent) [show]
.
Covalent bonds include disulfide bonds (-S-S-) between the side radicals of cysteines located in different parts of the polypeptide chain; isopeptide, or pseudopeptide, - between the amino groups of side radicals of lysine, arginine, and not α-amino groups, and COOH groups of side radicals of aspartic, glutamic and aminocitric acids, and not α-carboxyl groups of amino acids. Hence the name of this type of bond - peptide-like. A rare ester bond is formed by the COOH group of dicarboxylic amino acids (aspartic, glutamic) and the OH group of hydroxyamino acids (serine, threonine).
- weak (polar and van der Waals) [show]
.
TO polar bonds include hydrogen and ionic. Hydrogen bonds, as usual, occur between the -NH 2 , -OH or -SH group of the side radical of one amino acid and the carboxyl group of another. Ionic, or electrostatic, bonds are formed when the charged groups of side radicals -NH + 3 (lysine, arginine, histidine) and -COO - (aspartic and glutamic acids) come into contact.
Non-polar, or van der Waals, bonds are formed between hydrocarbon radicals amino acids. Hydrophobic radicals of the amino acids alanine, valine, isoleucine, methionine, phenylalanine interact with each other in an aqueous environment. Weak van der Waals bonds promote the formation of a hydrophobic core of nonpolar radicals inside the protein globule. The more nonpolar amino acids there are, the greater the role van der Waals bonds play in the folding of the polypeptide chain.
Numerous bonds between the side radicals of amino acids determine the spatial configuration of the protein molecule.
Features of the organization of protein tertiary structure . The conformation of the tertiary structure of the polypeptide chain is determined by the properties of the side radicals of the amino acids included in it (which do not have a noticeable effect on the formation of primary and secondary structures) and the microenvironment, i.e., the environment. When folded, the polypeptide chain of a protein tends to take on an energetically favorable form, characterized by a minimum of free energy. Therefore, nonpolar R-groups, “avoiding” water, form, as it were, the internal part of the tertiary structure of the protein, where the main part of the hydrophobic residues of the polypeptide chain is located. There are almost no water molecules in the center of the protein globule. The polar (hydrophilic) R groups of the amino acid are located outside this hydrophobic core and are surrounded by water molecules. The polypeptide chain is intricately bent in three-dimensional space. When it bends, the secondary helical conformation is disrupted. The chain “breaks” at weak points where proline or hydroxyproline are located, since these amino acids are more mobile in the chain, forming only one hydrogen bond with other peptide groups. Another bend site is glycine, which has a small R group (hydrogen). Therefore, the R-groups of other amino acids, when stacked, tend to occupy the free space at the location of glycine. A number of amino acids - alanine, leucine, glutamate, histidine - contribute to the preservation of stable helical structures in protein, and such as methionine, valine, isoleucine, aspartic acid favor the formation of β-structures. In a protein molecule with a tertiary configuration, there are regions in the form of α-helices (helical), β-structures (layered) and a random coil. Only the correct spatial arrangement of the protein makes it active; its violation leads to changes in protein properties and loss of biological activity.
Quaternary protein structure
Proteins consisting of one polypeptide chain have only tertiary structure. These include myoglobin - a muscle tissue protein involved in the binding of oxygen, a number of enzymes (lysozyme, pepsin, trypsin, etc.). However, some proteins are built from several polypeptide chains, each of which has a tertiary structure. For such proteins, the concept of quaternary structure has been introduced, which is the organization of several polypeptide chains with a tertiary structure into a single functional protein molecule. Such a protein with a quaternary structure is called an oligomer, and its polypeptide chains with a tertiary structure are called protomers or subunits (Fig. 4).

At the quaternary level of organization, proteins retain the basic configuration of the tertiary structure (globular or fibrillar). For example, hemoglobin is a protein with a quaternary structure and consists of four subunits. Each of the subunits is a globular protein and, in general, hemoglobin also has a globular configuration. Hair and wool proteins - keratins, related in tertiary structure to fibrillar proteins, have a fibrillar conformation and a quaternary structure.
Stabilization of protein quaternary structure . All proteins that have a quaternary structure are isolated in the form of individual macromolecules that do not break down into subunits. Contacts between the surfaces of subunits are possible only due to the polar groups of amino acid residues, since during the formation of the tertiary structure of each of the polypeptide chains, the side radicals of non-polar amino acids (which make up the majority of all proteinogenic amino acids) are hidden inside the subunit. Numerous ionic (salt), hydrogen, and in some cases disulfide bonds are formed between their polar groups, which firmly hold the subunits in the form of an organized complex. The use of substances that break hydrogen bonds or substances that reduce disulfide bridges causes disaggregation of protomers and destruction of the quaternary structure of the protein. In table 1 summarizes the data on the bonds that stabilize different levels of organization of the protein molecule [show] .
| Table 1. Characteristics of bonds involved in the structural organization of proteins | ||
| Organization level | Types of bonds (by strength) | Type of communication |
| Primary (linear polypeptide chain) | Covalent (strong) | Peptide - between the α-amino and α-carboxyl groups of amino acids |
| Secondary (α-helix, β-structures) | Weak | Hydrogen - between peptide groups (every first and fourth) of one polypeptide chain or between peptide groups of adjacent polypeptide chains |
| Covalent (strong) | Disulfide - disulfide loops within a linear region of a polypeptide chain | |
| Tertiary (globular, fibrillar) | Covalent (strong) | Disulfide, isopeptide, ester - between the side radicals of amino acids of different parts of the polypeptide chain |
| Weak | Hydrogen - between the side radicals of amino acids of different parts of the polypeptide chain Ionic (salt) - between oppositely charged groups of side radicals of amino acids of the polypeptide chain Van der Waals - between non-polar side radicals of amino acids of the polypeptide chain |
|
| Quaternary (globular, fibrillar) | Weak | Ionic - between oppositely charged groups of side radicals of amino acids of each of the subunits Hydrogen - between the side radicals of amino acid residues located on the surface of the contacting areas of the subunits |
| Covalent (strong) | Disulfide - between cysteine residues of each of the contacting surfaces of different subunits | |
Features of the structural organization of some fibrillar proteins
The structural organization of fibrillar proteins has a number of features compared to globular proteins. These features can be seen in the example of keratin, fibroin and collagen. Keratins exist in α- and β-conformations. α-Keratins and fibroin have a layered-folded secondary structure, however, in keratin the chains are parallel, and in fibroin they are antiparallel (see Fig. 3); In addition, keratin contains interchain disulfide bonds, while fibroin does not have them. Breakage of disulfide bonds leads to separation of polypeptide chains in keratins. On the contrary, education maximum number disulfide bonds in keratins through the action of oxidizing agents creates a strong spatial structure. In general, in fibrillar proteins, unlike globular ones, it is sometimes difficult to strictly distinguish between different levels of organization. If we accept (as for a globular protein) that the tertiary structure should be formed by laying one polypeptide chain in space, and the quaternary structure by several chains, then in fibrillar proteins several polypeptide chains are already involved in the formation of the secondary structure. A typical example of a fibrillar protein is collagen, which is one of the most abundant proteins in the human body (about 1/3 of the mass of all proteins). It is found in tissues that have high strength and low extensibility (bones, tendons, skin, teeth, etc.). In collagen, a third of the amino acid residues are glycine, and about a quarter or slightly more are proline or hydroxyproline.

The isolated polypeptide chain of collagen (primary structure) is similar to broken line. It contains about 1000 amino acids and has a molecular weight of about 10 5 (Fig. 5, a, b). A polypeptide chain is made up of a repeating trio of amino acids (triplet) next line-up: gly-A-B, where A and B are any amino acids other than glycine (most often proline and hydroxyproline). Collagen polypeptide chains (or α-chains) during the formation of secondary and tertiary structures (Fig. 5, c and d) cannot produce typical α-helices with helical symmetry. Proline, hydroxyproline and glycine (antihelical amino acids) interfere with this. Therefore, three α-chains form, as it were, twisted spirals, like three threads wrapping around a cylinder. Three helical α chains form a repeating collagen structure called tropocollagen (Fig. 5d). Tropocollagen in its organization is the tertiary structure of collagen. The flat rings of proline and hydroxyproline regularly alternating along the chain give it rigidity, as do the interchain bonds between the α-chains of tropocollagen (which is why collagen is resistant to stretching). Tropocollagen is essentially a subunit of collagen fibrils. The laying of tropocollagen subunits into the quaternary structure of collagen occurs in a stepwise manner (Fig. 5e).
Stabilization of collagen structures occurs due to interchain hydrogen, ionic and van der Waals bonds and a small number of covalent bonds.
The α-chains of collagen have different chemical structures. There are α 1 chains different types(I, II, III, IV) and α 2 chains. Depending on which α 1 - and α 2 -chains are involved in the formation of the three-stranded helix of tropocollagen, four types of collagen are distinguished:
- the first type - two α 1 (I) and one α 2 chain;
- the second type - three α 1 (II) chains;
- third type - three α 1 (III) chains;
- fourth type - three α 1 (IV) chains.
The most common collagen is the first type: it is found in bone tissue, skin, tendons; Type II collagen is found in cartilage tissue etc. There can be different types of collagen in one type of tissue.
The ordered aggregation of collagen structures, their rigidity and inertness ensure the high strength of collagen fibers. Collagen proteins also contain carbohydrate components, i.e. they are protein-carbohydrate complexes.
Collagen is an extracellular protein that is formed by connective tissue cells found in all organs. Therefore, with damage to collagen (or disruption of its formation), multiple violations of the supporting functions of the connective tissue of organs occur.
| Page 3 | total pages: 7 |
A linear polypeptide chain in water is capable of spontaneously folding into a complex three-dimensional structure (globule), and only in this folded form can proteins perform chemical catalysis and other interesting work. Therefore, it is fundamentally important for us to know the three-dimensional folding of the protein, since only at this level it becomes clear how the protein works.
Question: How many three-dimensional structures correspond to a particular protein?
Answer: One, up to slight mobility of small “disordered” loops. There is exactly one known exception, when one sequence corresponds to 2 quite different structures, these are prions.
Question: What is the three-dimensional structure of a protein based on?
Answer: in short, mainly on a large number of non-covalent interactions. In principle, the chemical groups of a protein can form: (1) a hydrogen bond, these groups are present both in the main chain and in some side groups, (2) an ionic bond - electrostatic interaction between oppositely charged side groups, (3) Van der Waals interaction and (4) the hydrophobic effect on which the overall structure of the protein rests. The bottom line is that a protein always contains hydrophobic aromatic residues; it is energetically unfavorable for them to come into contact with polar water molecules, but it is advantageous for them to “stick together” with each other. Thus, when a protein folds, hydrophobic groups are pushed out of the aqueous environment, “sticking” to each other and forming a “hydrophobic core,” while polar and charged groups, on the contrary, tend to the aqueous environment, forming the surface of the protein globule. Also (5) the side groups of two cysteine residues can form a disulfide bridge between themselves - a full-fledged covalent bond that rigidly fixes the protein.
Accordingly, all amino acids are divided into hydrophobic, polar (hydrophilic), positively and negatively charged. Plus cysteines, which can form covalent bonds with each other. Glycine has special properties - it does not have a side group, which greatly limits the conformational mobility of other residues, so it can “bend” very strongly and is located in places where protein chain need to be deployed. In proline, on the contrary, the side group forms a ring covalently bound to the main chain, rigidly fixing its conformation. Prolines are found where it is necessary to make the protein chain rigid and inflexible. Many diseases are associated with a mutation from proline to glycine, which causes the protein structure to “float” slightly.
Question: How do we even know about the three-dimensional structures of proteins?
Answer: from the experiment, this is absolutely reliable data.
Now there are 3 methods for experimental determination of protein structure: nuclear magnetic resonance (NMR), cryo-EM (electron microscopy) and X-ray diffraction analysis of protein crystals.
NMR can determine the structure of a protein in solution, but it only works for very small proteins (it is impossible to deconvolute for large ones). 
This method was important for the general proof that a protein has only one three-dimensional structure and that the structure of the protein in crystal is identical to that in solution. This is a very expensive method, since it requires isotopically tagged proteins.
Cryo-EM involves simply freezing a protein solution and microscopying it. The disadvantage of the method is low resolution (only general shape molecule, but it is not visible how it is arranged inside), plus the density of the protein is close to the density of water/solvent, so the signal is drowned in a high level of noise. This method actively uses computer technology working with pictures and statistics to extract signal from noise. 
Millions of pictures of protein molecules are selected, divided into classes depending on the orientation of the molecule relative to the substrate, averaging across classes, generation of eigenimages, a new round of averaging, and so on until it converges. Then from the information from different classes it is possible to reconstruct a low-resolution 3D view of the molecule. If there is internal symmetry of particles (for example, in cryo-EM analysis of viruses), then each particle can be averaged in accordance with symmetry operators - then the resolution will be even better, but worse than in the case of X-ray diffraction analysis.
X-ray diffraction analysis is the main method for determining protein structures. The main advantage is that it is potentially possible to obtain crystals of even very large complexes from many dozens of proteins (for example, this is how the structure of the ribosome was determined - Nobel Prize 2009). The disadvantage of this method is that you first need to obtain a protein crystal, but not every protein wants to crystallize. 
But after the crystal is obtained, by diffraction x-ray radiation can unambiguously determine the positions of all (ordered) atoms in a protein molecule, this method gives the most high resolution and allows for best cases see the positions of individual atoms. It was proven that the structure of the protein in crystal uniquely corresponds to the structure in solution.
Now there is a convention - if you have determined the structure of a protein using any of the experimental physical methods, the structure should be placed in the public domain in the Protein Data Bank (PDB, www.pdb.org), currently there are more than 90,000 structures there (however, many of them are repeating, for example, complexes of the same same protein with various small molecules, such as drugs). In PDB, all structures are in a standard format called, suddenly, pdb. This is a text format in which each atom of the structure corresponds to one line, which indicates the number of the atom in the structure, the name of the atom (carbon, nitrogen, etc.), the name of the amino acid that the atom is part of, the name of the protein chain (A, B, C, etc. , if this is a crystal of a complex of several proteins), the number of the amino acid in the chain and the three-dimensional coordinates of the atom in angstroms relative to the origin, plus the so-called temperature factor and population (these are purely crystallographic parameters).
ATOM 1 N HIS A 17 -12.690 8.753 5.446 1.00 29.32 N ATOM 2 CA HIS A 17 -11.570 8.953 6.350 1.00 21.61 C ATOM 3 C HIS A 17 -10.274 8.970 5.544 1.00 22. 01 C ATOM 4 O HIS A 17 -10.193 8.315 4.491 1.00 29.95 O ATOM 5 CB HIS A 17 -11.462 7.820 7.380 1.00 23.64 C ATOM 6 CG HIS A 17 -12.551 7.811 8.421 1.00 21.18 C ATOM 7 ND1 HIS A 17 -13.731 7.137 8.1 94 1.00 28.94 N ATOM 8 CD2 HIS A 17 -12.634 8.384 9.644 1.00 21.69 C ATOM 9 CE1 HIS A 17 -14.492 7.301 9.267 1.00 27.01 C ATOM 10 NE2 HIS A 17 -13.869 8.058 10.168 1.00 22.66 N ATOM 11 N ILE A 18 -9.2 69 9.660 6.089 1.00 19.45 N ATOM 12 CA ILE A 18 - 7.910 9.377 5.605 1.00 18.67 C ATOM 13 C ILE A 18 -7.122 8.759 6.749 1.00 16.24 C ATOM 14 O ILE A 18 -7.425 8.919 7.929 1.00 18.80 O ATOM 15 CB ILE A 18 -7.228 10.640 5.088 1.00 20.22 C ATOM 16 CG1 ILE A 18 -7.062 11.686 6.183 1.00 18.52 C ATOM 17 CG2 ILE A 18 -7.981 11.176 3.889 1.00 24.61 C ATOM 18 CD1 ILE A 18 -6.161 12.824 5.749 1.00 28.21 C ATOM 19 N ASN A 19 -6.121 8.023 6.349 1.00 15.46 N ATOM 20 CA ASN A 19 -5.239 7.306 7.243 1.00 14.34 C ATOM 21 C ASN A 19 -4.012 8.178 7.507 1.00 14.83 C ATOM 22 O ASN A 19 -3.431 8.715 6.575 1.00 18.03 O ATOM 23 CB ASN A 19 -4.825 6.003 6.573 1.00 17.71 C ATOM 24 CG ASN A 19 -6.062 5.099 6.413 1.00 21.26 C ATOM 25 OD1 ASN A 19 -6.606 4.651 7.400 1.00 26.18 O ATOM 26 ND2 ASN A 19 -6.320 4.899 5.151 1.00 31.73N
Next there is special programs, which, according to data from this text file, can graphically display the beautiful three-dimensional structure of a protein molecule, which can be rotated on the monitor screen and, as Guy Dodson said, “touch the molecule with the mouse” (for example, PyMol, CCP4mg, old RasMol). That is, it’s easy to look at protein structures - install the program, download the desired structure from PDB and enjoy the beauty of nature.
4. Analyze the structure
So, we understand the basic idea: a protein is a linear polymer that folds in an aqueous solution under the influence of many weak interactions into a stable and unique three-dimensional structure for a given protein, and in this form is capable of performing its function. There are several levels of organization of protein structures. Above, we have already become acquainted with the primary structure - a linear sequence of amino acids that can be written down on a line.
The secondary structure of a protein is determined by the interactions of the atoms of the protein backbone. As mentioned above, the main chain of a protein includes hydrogen bond donors and acceptors, thus the main chain can acquire some structure. More precisely, several different structures (the details still depend on the different side groups), since the formation of different alternative hydrogen bonds between the groups of the main chain is possible. The structures are as follows: alpha helix, beta sheets (consisting of several beta strands), which can be parallel or anti-parallel, beta turn. Plus, part of the chain may not have a pronounced structure, for example, in the region of the protein loop turn. These types of structures have their own established schematic designations - alpha helix in the form of a helix or cylinder, beta strands in the form of wide arrows. The secondary structure can be predicted quite reliably from the primary structure (JPred is the standard), alpha helices are predicted most accurately, and there are overlaps with beta strands.
The tertiary structure of a protein is determined by the interaction of side groups of amino acid residues; this is the three-dimensional structure of the protein. One can imagine that the secondary structure has been formed and now these helices and beta strands want to fit together into a compact three-dimensional structure, so that all the hydrophobic side groups quietly “stick together” in the depths of the protein globule, forming a hydrophobic core, and the polar and charged residues stick out out into the water, forming the surface of the protein and stabilizing the contacts between the elements of the secondary structure. The tertiary structure is depicted schematically in several ways. If you just draw all the atoms, you'll get a mess (although when we analyze the active site of a protein, we want to look at all the atoms of the active residues). 
If we want to see how the whole protein is organized in general, we can display only some of the atoms of the main chain to see its progress. As an option, you can draw a beautiful diagram, where elements of the secondary structure are schematically drawn on top of the actual arrangement of atoms - this way the protein folding is visible at first glance. After studying the entire structure in a general, schematic form, you can display the chemical groups of the active site and focus on them. The problem of predicting the tertiary structure of a protein is nontrivial and cannot be solved in the general case, although it can be solved in special cases. More details below.
Quaternary protein structure - yes, there is such a thing, although not all proteins have it. Many proteins work on their own (monomers, in this case a monomer means a single folded polypeptide chain, that is, the entire protein), then their quaternary structure is equal to the tertiary one. However, quite a lot of proteins work only in a complex consisting of several polypeptide chains (subunits or monomers - dimers, trimers, tetramers, multimers), then such an assembly of several individual chains is called a quaternary structure. The most banal example is hemoglobin, consisting of 4 subunits; the most beautiful example, in my opinion, is the bacterial protein TRAP, consisting of 11 identical subunits.
5. Computational tasks
A protein is a complex system of thousands of atoms, so without the use of computers it is impossible to understand the structure of a protein. There are many problems, both solved at an acceptable level and not solved at all. I will list the most relevant ones:At the level of the primary structure– searching for proteins with similar amino acid sequences, building evolutionary trees based on them, etc. – classical problems bioinformatics. The main hub is NCBI - The National Center for Biotechnology Information, www.ncbi.nlm.nih.gov. To search for proteins with similar sequences, BLAST is standardly used: blast.ncbi.nlm.nih.gov/Blast.cgi
Prediction of protein solubility. The point is that if we read the genome of an animal, determine the protein sequences from it, and clone these genes into Escherichia coli or the baculovirus expression system, it turns out that when expressed in these systems, approximately a third of the proteins will not fold into the correct structure , and, as a result, will be insoluble. Here it turns out that large proteins actually consist of separate “domains”, each of which represents an autonomous, functional part of the protein (carrying one of its functions) and often by “cutting out” a separate domain from a gene, you can obtain a soluble protein and determine its structure and conduct experiments with it. People are trying to use machine learning ( neural networks, SVM and other classifiers) to predict protein solubility, but it works quite poorly (Google will show a lot of things for the query “protein solubility prediction” - there are many servers, but in my experience they all work disgustingly on my proteins). Ideally, I would like to see a service that would reliably tell where those soluble domains are located in a protein, so that they can be cut out and worked with - there is no such service.
At the secondary structure level– prediction of the same secondary structure from the primary one (JPred)
At the tertiary structure level– search for proteins with similar three-dimensional structures (DALI, en.wikipedia.org/wiki/Structural_alignment),
Search for structures based on a given sub-structure. For example, I have the arrangement of three active site amino acids in space. I want to find structures that contain the same three amino acids in the same relative arrangement, or find protein structures, the mutation of which will make it possible to arrange the necessary amino acids in the desired way. (Google “protein substructure search”)
Prediction of potential mobility of a three-dimensional structure, possible conformational changes - normal mode analysis, ElNemo.
At the level of quaternary structure– suppose the structures of two proteins are known. They are known to form a complex. Predict the structure of the complex (determine how these two proteins will interact through shape matching, for example). Google “protein-protein docking”
6. Protein structure prediction
I highlighted this computational problem in a separate section, because it is large, fundamental and cannot be solved in the general case.We know experimentally that if you take a protein, completely unfold it and throw it into water, it will fold back into its original state in a time of milliseconds to seconds (this statement is true at least for small globular proteins without any pathologies). This means that all the information necessary to determine the three-dimensional structure of a protein is implicitly contained in its primary sequence, which is why there is a great desire to learn how to predict the three-dimensional structure of a protein from the amino acid sequence in silico! However, this problem has not yet been solved in the general case. What's the matter? The fact is that the primary sequence does not explicitly contain the information necessary to construct the structure. Firstly, there is no information about the conformation of the main chain - and it has significant mobility, although somewhat limited for steric reasons. Plus, each side chain of each amino acid can be in different conformations; for long side chains like arginine, this can be more than a dozen conformations.
What to do? There is one quite well known to Khabra residents general approach, called “molecular dynamics” and suitable for any molecules and systems. We take an unfolded protein, assign random velocities to all atoms, count the interactions between the atoms, and repeat until the system reaches a stable state corresponding to the folded protein. Why doesn't this work? Because modern computing power makes it possible, over months of cluster operation, to count tens of nanoseconds for a system of thousands of atoms, such as a protein placed in water. The protein folding time is milliseconds or more, that is, there is not enough computing power, the gap is several orders of magnitude. However, a couple of years ago the Americans made some breakthrough. They used special hardware optimized for vector calculations and after optimization at the hardware level, over months of machine operation they were able to calculate the molecular dynamics down to milliseconds for a very small protein and the protein folded, the structure corresponded to the experimentally determined one (http://en.wikipedia.org/wiki /Anton_(computer))! However, it is too early to celebrate victory. They took a very small (its size is 5-10 times smaller than the average protein) and one of the fastest folding proteins, a classic model protein on which folding was studied. For large proteins, the calculation time increases nonlinearly and will take years, which means there is still work to be done.
A different approach is implemented in Rosetta. They break the protein sequence into very short (3-9 residues) fragments and look at what conformations for these fragments are present in the PDB, then run Monte Carlo on all the variants and see what happens. Sometimes something good comes out, but in my cases, after a few days of cluster operation, you get such a donut that a silent question arises: “Who wrote their evaluation function that puts some kind of good grade this squiggle?
There are also tools for manual modeling - you can predict the secondary structure and try to manually twist it, finding the best fit. Some brilliant people They even released a toy FoldIt, which represents the protein schematically and allows you to fold it, as if assembling a puzzle (I recommend it for those interested in structure!). There is a completely official competition for protein structure predictors called CASP. The point is that when experimenters determine new structure protein that has no analogues in the PDB, they may not immediately publish it in the PDB, but submit the sequence of this protein to the CASP prediction competition. After a while, when everyone has finished their predictive models, the experimenters lay out their experimentally determined protein structure and see how well the predictors worked. The most interesting thing is that FoldIt players, who were not scientists, somehow won CASP against protein structure modeling professionals and predicted the protein structure more accurately. However, even these successes do not allow us to say that the problem of predicting protein structure is being solved - very often the model is very far from the real structure.
All this related to protein modeling ab initio, when there is no a priori information about the structure. However, very often there are situations when for some protein a distant relative with an already known structure is present in the PDB. By relative is meant a protein with a similar primary sequence. Proteins with a primary sequence similarity greater than 30% are considered to have identical backbone folding (although similar folding has also been observed for proteins that do not exhibit any statistically significant primary sequence similarity). If there is a homologue (similar protein) with a known structure, you can do “homologous modeling”, that is, simply “stretch” the sequence of your protein onto known structure homologue, and then drive energy minimization in order to somehow sort this whole thing out. This modeling shows good results In the presence of very close homologues, the further away the homologue is, the greater the error. Tools for homology modeling – Modeller, SwissModel.
You can solve other problems, for example, try to simulate what will happen if you introduce one or another mutation into a protein. For example, if you replace a hydrophilic amino acid on the surface of a protein with another hydrophilic one, then most likely the structure of the protein will not change at all. If you replace an amino acid from a hydrophobic core with another hydrophobic one, but of a different size, then most likely the protein fold will remain the same, but will “shift” slightly by fractions of an angstrom. If you replace an amino acid from a hydrophobic core with a charged one, then most likely the protein will simply “explode” and will not be able to fold.
It may seem like things aren't so bad and we have a pretty good understanding of protein folding. Yes, we understand some things, for example, to some extent we understand the general physical principles, underlying the folding of the polypeptide chain - they are discussed in the wonderful textbook by Ptitsyn and Finkelstein “Physics of Protein”. However, this general understanding does not allow us to answer the questions “Will this protein fold or not?”, “What structure will this protein have?”, “How to make a protein with the desired structure?”
Here is one illustration: we want to localize one of the domains of a large protein, this standard task. We have a fragment that folds and is soluble, meaning it is a living and healthy protein. We want to find its minimal part and start using the methods genetic engineering remove 2-3 amino acids from both ends, express such a trimmed protein in bacteria and observe its folding experimentally. We make dozens of constructs with such small deletions and see the following picture: a completely soluble and living protein differs from a completely dead and non-folding protein by 3 amino acids. I repeat, this is an objective experimental result. The problem is that there is currently no computational method that would predict the folding of a protein at least on a yes/no level and tell me where the boundary between a folding and a nonfolding protein is, so we are forced to clone and experimentally test dozens of variants. This is just one illustration of the fact that our understanding of protein structure is far from perfect. As Richard Feynman said, “What I cannot recreate, I do not understand.”
So, gentlemen, programmers, physicists and mathematicians, we still have work to do.
On this optimistic note, allow me to take my leave, thank you to everyone who mastered this opus.
For a deep understanding of the subject area, I recommend the following minimum:
1) “Physics of Protein” Ptitsyn and Finkelstein. Alexey Vitalievich Finkelshtein posted most of the material online, which I gratefully recommend using: phys.protres.ru/lectures/protein_physics/index.html (and I stole a few pictures from there)
2) Patrushev, “Artificial genetic systems,” especially part II “Protein engineering.” Available on torrents in Djvu format
3) For information published in biological scientific journals, there is the official search engine PubMed (www.pubmed.org) - it’s worth asking him to read about “protein engineering” and the like.
Tags:
- biology
- bioinformatics
- biotechnology
L Due to the interaction of functional groups of amino acids, linear polypeptide chains of individual proteins acquire a certain spatial three-dimensional structure, called “conformation”. All molecules of individual proteins (i.e., having the same primary structure) form the same conformation in solution. Consequently, all the information necessary for the formation of spatial structures is located in the primary structure of proteins.
In proteins, there are 2 main types of conformation of polypeptide chains: secondary and tertiary structures.
2. Secondary structure of proteins - spatial structure resulting from the interaction between functional groups of the peptide backbone.
In this case, peptide chains can acquire regular structures of two types: α-helices
β-structure By β-structure we mean a figure similar to a sheet folded like an accordion. The figure is formed due to the formation of many hydrogen bonds between the atoms of the peptide groups of the linear regions of one polypeptide chain making bends, or between different polypeptide groups.

Bonds are hydrogen, they stabilize individual fragments of macromolecules.
3. Tertiary structure of proteins - a three-dimensional spatial structure formed due to interactions between amino acid radicals, which can be located at a considerable distance from each other in the polypeptide chain.
Structurally consists of elements of secondary structure, stabilized by various types of interactions, in which hydrophobic interactions play a critical role
stabilization of the tertiary structure of the protein takes part:
· covalent bonds (between two cysteine residues - disulfide bridges);
· ionic bonds between oppositely charged side groups of amino acid residues;
· hydrogen bonds;
· hydrophilic-hydrophobic interactions. When interacting with surrounding water molecules, the protein molecule “tends” to fold so that the nonpolar side groups of amino acids are isolated from the aqueous solution; polar hydrophilic side groups appear on the surface of the molecule.
4. Quaternary structure is the relative arrangement of several polypeptide chains within a single protein complex. Protein molecules that make up a protein with a quaternary structure are formed separately on ribosomes and only after completion of synthesis form a common supramolecular structure. A protein with a quaternary structure can contain both identical and different polypeptide chains. Participate in the stabilization of the quaternary structure the same types of interactions as in the stabilization of tertiary. Supramolecular protein complexes can consist of dozens of molecules.
Role.
The formation of peptides in the body occurs within a few minutes, while chemical synthesis in the laboratory is quite long process, which can take several days, and the development of synthesis technology can take several years. However, despite this, there are quite strong arguments in favor of carrying out work on the synthesis of analogues of natural peptides. First, by chemical modification of peptides it is possible to confirm the primary structure hypothesis. The amino acid sequences of some hormones became known precisely through the synthesis of their analogues in the laboratory.
Secondly, synthetic peptides allow us to study in more detail the relationship between the structure of an amino acid sequence and its activity. To clarify the relationship between the specific structure of the peptide and its biological activity, a huge amount of work was carried out on the synthesis of more than one thousand analogues. As a result, it was found that replacing just one amino acid in the structure of a peptide can increase its biological activity several times or change its direction. And changing the length of the amino acid sequence helps determine the location of the active centers of the peptide and the site of receptor interaction.
Thirdly, thanks to the modification of the original amino acid sequence, it became possible to obtain pharmacological drugs. The creation of analogues of natural peptides makes it possible to identify more “effective” configurations of molecules that enhance biological effect or make it last longer.
Fourthly, chemical synthesis of peptides is economically beneficial. Most therapeutic drugs would cost tens of times more if they were made from a natural product.
Often, active peptides are found in nature only in nanogram quantities. Plus, methods for purifying and isolating peptides from natural sources cannot completely separate the desired amino acid sequence from peptides of the opposite or other effect. And in the case of specific peptides synthesized by the human body, they can only be obtained through synthesis in laboratory conditions.
57. Classification of proteins: simple and complex, globular and fibrillar, monomeric and oligomeric. Functions of proteins in the body.
Classification by type of structure
By general type The structure of proteins can be divided into three groups:
1. Fibrillar proteins - form polymers, their structure is usually highly regular and is maintained mainly by interactions between different chains. They form microfilaments, microtubules, fibrils, and support the structure of cells and tissues. Fibrillar proteins include keratin and collagen.
2. Globular proteins are water soluble, the general shape of the molecule is more or less spherical.
3. Membrane proteins - have domains that cross the cell membrane, but parts of them protrude from the membrane into the intercellular environment and the cytoplasm of the cell. Membrane proteins function as receptors, that is, they transmit signals and also provide transmembrane transport of various substances. Transporter proteins are specific; each of them allows only certain molecules or a certain type of signal to pass through the membrane.
Simple proteins , Complex proteins
In addition to peptide chains, many proteins also contain non-amino acid groups, and according to this criterion, proteins are divided into two large groups - simple and complex proteins(proteids). Simple proteins consist only of polypeptide chains; complex proteins also contain non-amino acid, or prosthetic, groups.
Simple.
Among the globular proteins we can distinguish:
1. albumins - soluble in water over a wide pH range (from 4 to 8.5), precipitated with a 70-100% solution of ammonium sulfate;
2. polyfunctional globulins with a higher molecular weight, less soluble in water, soluble in saline solutions, often contain a carbohydrate part;
3. histones are low molecular weight proteins with a high content of arginine and lysine residues in the molecule, which determines their basic properties;
4. protamines are distinguished by an even higher arginine content (up to 85%), like histones, they form stable associates with nucleic acids, acting as regulatory and repressor proteins - an integral part of nucleoproteins;
5. prolamines are characterized by a high content of glutamic acid (30-45%) and proline (up to 15%), insoluble in water, soluble in 50-90% ethanol;
6. Glutelins contain about 45% glutamic acid, like prolamins, and are often found in cereal proteins.
Fibrillar proteins are characterized by a fibrous structure and are practically insoluble in water and saline solutions. Polypeptide chains in molecules are located parallel to one another. Participate in the formation of structural elements of connective tissue (collagens, keratins, elastins).
Complex proteins
(proteids, holoproteins) are two-component proteins that, in addition to peptide chains (simple protein), contain a non-amino acid component - a prosthetic group. When complex proteins are hydrolyzed, in addition to amino acids, the non-protein part or its breakdown products are released.
Various organic (lipids, carbohydrates) and inorganic (metals) substances can act as a prosthetic group.
Depending on the chemical nature of the prosthetic groups, the following classes are distinguished among complex proteins:
· Glycoproteins containing covalently bound carbohydrate residues as a prosthetic group and their subclass - proteoglycans, with mucopolysaccharide prosthetic groups. The formation of bonds with carbohydrate residues usually involves hydroxyl groups serine or threonine. Most extracellular proteins, in particular immunoglobulins, are glycoproteins. The carbohydrate part of proteoglycans is ~95%; they are the main component of the intercellular matrix.
· Lipoproteins containing non-covalently bound lipids as a prosthetic part. Lipoproteins are formed by apolipoprotein proteins that bind lipids to them and perform the function of lipid transport.
· Metalloproteins containing non-heme coordinated metal ions. Among metalloproteins there are proteins that perform storage and transport functions (for example, iron-containing ferritin and transferrin) and enzymes (for example, zinc-containing carbonic anhydrase and various superoxide dismutases containing copper, manganese, iron and other metal ions as active centers)
· Nucleoproteins containing non-covalently bound DNA or RNA, in particular, chromatin, which makes up chromosomes, is a nucleoprotein.
· Phosphoproteins containing covalently bound phosphoric acid residues as a prosthetic group. Hydroxyl groups of serine or threonine participate in the formation of an ester bond with phosphate; milk casein, in particular, is a phosphoprotein:
· Chromoproteins are the collective name for complex proteins with colored prosthetic groups of various chemical natures. These include many proteins with a metal-containing porphyrin prosthetic group that perform various functions - hemoproteins (proteins containing heme as a prosthetic group - hemoglobin, cytochromes, etc.), chlorophylls; flavoproteins with a flavin group, etc.
1. Structural function
2. Protective function
3. Regulatory function
4. Alarm function
5. Transport function
6. Spare (backup) function
7. Receptor function
8. Motor (motor) function
Proteins are organic high-molecular compounds. These substances are also called proteins and polypeptides. Next, let's look at the structure and functions of proteins.
General information
The chemical structure of proteins is represented by alpha amino acids connected in a chain through a peptide bond. In living organisms, the composition is determined by the genetic code. In the synthesis process, in most cases, 20 amino acids of the standard type are used. Their many combinations form protein molecules with a wide variety of properties. Amino acid residues are often subject to post-translational modifications. They can arise before the protein begins to perform its functions, and during its activity in the cell. In living organisms, several molecules often form complex complexes. An example is photosynthetic association.
Purpose of connections
Proteins are considered an important component of human and animal nutrition due to the fact that their bodies cannot synthesize all the necessary amino acids. Some of them should come with protein foods. The main sources of compounds are meat, nuts, milk, fish, and grains. To a lesser extent, proteins are present in vegetables, mushrooms and berries. During digestion through enzymes, consumed proteins are broken down into amino acids. They are already used in the biosynthesis of their own proteins in the body or undergo further breakdown to obtain energy.

Historical background
The sequence of the insulin protein structure was first determined by Frederij Senger. For his work he received the Nobel Prize in 1958. Sanger used the sequencing method. Using X-ray diffraction, three-dimensional structures of myoglobin and hemoglobin were subsequently obtained (in the late 1950s). The work was carried out by John Kendrew and Max Perutz.
Protein molecule structure
It includes linear polymers. They, in turn, consist of alpha amino acid residues, which are monomers. In addition, the protein structure may include components of a non-amino acid nature and modified amino acid residues. When designating components, 1- or 3-letter abbreviations are used. A compound containing from two to several dozen residues is often referred to as a “polypeptide.” As a result of the interaction of the alpha-carboxyl group of one amino acid with the alpha-amino group of another, bonds appear (during the formation of the protein structure). The C- and N-terminal ends of the compound are distinguished, depending on which group of the amino acid residue is free: -COOH or -NH 2 . In the process of protein synthesis on the ribosome, the first terminal residue is usually a methionine residue; the subsequent ones are attached to the C-terminus of the previous ones.

Levels of organization
They were proposed by Lindrem-Lang. Despite the fact that this division is considered somewhat outdated, it is still used. It was proposed to distinguish four levels of connection organization. The primary structure of a protein molecule is determined by the genetic code and characteristics of the gene. Higher levels are characterized by formation during protein folding. The spatial structure of a protein is determined as a whole by the amino acid chain. Nevertheless, it is quite labile. She may be influenced external factors. In this regard, it is more correct to talk about the conformation of the compound that is the most favorable and energetically preferable.
Level 1
It is represented by a sequence of amino acid residues of a polypeptide chain. As a rule, it is described using one- or three-letter notations. The primary structure of proteins is characterized by stable combinations of amino acid residues. They perform specific tasks. Such “conservative motifs” remain preserved during species evolution. They can often be used to predict the problem of an unknown protein. Assessing the degree of similarity (homology) in amino acid chains from various organisms, it is possible to determine the evolutionary distance formed between the taxa that make up these organisms. The primary structure of proteins is determined by sequencing or by the original complex of its mRNA using a genetic code table.

Local ordering of a chain section
This is the next level of organization - the secondary structure of proteins. There are several types of it. Local ordering of a portion of a polypeptide chain is stabilized by hydrogen bonds. The most popular types are:

Spatial structure
The tertiary structure of proteins includes elements of the previous level. They are stabilizing different types interactions. Hydrophobic bonds are of paramount importance. Stabilization involves:
- Covalent interactions.
- Ionic bonds formed between amino acid side groups that have opposite charges.
- Hydrogen interactions.
- Hydrophobic bonds. In the process of interaction with surrounding elements H 2 O, the protein folds so that the side non-polar amino acid groups are isolated from the aqueous solution. Hydrophilic groups (polar) appear on the surface of the molecule.
The tertiary structure of proteins is determined by magnetic (nuclear) resonance methods, certain types of microscopy and other methods.
Laying principle
Research has shown that it is convenient to identify one more level between levels 2 and 3. It is called “architecture”, “laying motif”. It is determined by the relative position of the components of the secondary structure (beta strands and alpha helices) within the boundaries of a compact globule - the protein domain. It can exist independently or be included in a larger protein along with other similar proteins. It has been established that the styling motives are quite conservative. They are found in proteins that have neither evolutionary nor functional relationships. The definition of architecture is the basis of rational (physical) classification.

Domain organization
At relative position Several chains of polypeptides within one protein complex form the quaternary structure of proteins. The elements that make up it are formed separately on ribosomes. Only upon completion of synthesis does this protein structure begin to form. It can contain both different and identical polypeptide chains. The quaternary structure of proteins is stabilized due to the same interactions as at the previous level. Some complexes may include several dozen proteins.
Protein structure: protective tasks
Polypeptides of the cytoskeleton, acting in some way as reinforcement, give many organelles their shape and participate in its change. Structural proteins provide protection for the body. For example, collagen is such a protein. It forms the basis in the intercellular substance of connective tissues. Keratin also has a protective function. It forms the basis of horns, feathers, hair and other derivatives of the epidermis. When proteins bind toxins, in many cases detoxification of the latter occurs. This is how the task of chemical protection of the body is accomplished. Particularly important role in the process of neutralizing toxins in human body liver enzymes play. They are able to break down poisons or convert them into soluble form. This facilitates faster transport from the body. Proteins present in blood and other body fluids provide immune defense, triggering a response to both pathogen attack and injury. Immunoglobulins (antibodies and components of the complement system) are able to neutralize bacteria, foreign proteins and viruses.
Regulatory mechanism
Protein molecules, which act neither as an energy source nor as a building material, control many intracellular processes. Thus, due to them, translation, transcription, slicing, and the activity of other polypeptides are regulated. The regulatory mechanism is based on enzymatic activity or manifests itself due to specific binding to other molecules. For example, transcription factors, activator polypeptides, and repressor proteins are capable of controlling the intensity of gene transcription. In doing so, they interact with gene regulatory sequences. The most important role in controlling the course of intracellular processes is assigned to protein phosphatases and protein kinases. These enzymes trigger or inhibit the activity of other proteins by adding or removing phosphate groups from them.

Signal task
It is often combined with the regulatory function. This is due to the fact that many intracellular, as well as extracellular, polypeptides can transmit signals. Growth factors, cytokines, hormones and other compounds have this ability. Steroids are transported through the blood. The interaction of the hormone with the receptor acts as a signal that triggers the cell response. Steroids control the content of compounds in the blood and cells, reproduction, growth and other processes. An example is insulin. It regulates glucose levels. The interaction of cells is carried out through signal protein compounds transmitted through the intercellular substance.
Transport of elements
Soluble proteins involved in the movement of small molecules have a high affinity for the substrate, which is present in increased concentration. They also have the ability to release it easily in areas where its content is low. An example is the transport protein hemoglobin. It moves oxygen from the lungs to other tissues, and from them it transfers carbon dioxide. Some membrane proteins are also involved in the transport of small molecules through cell walls, changing them. The lipid layer of the cytoplasm is waterproof. This prevents the diffusion of charged or polar molecules. Membrane transport connections are usually divided into carriers and channels.
Backup connections
These proteins form so-called reserves. They accumulate, for example, in plant seeds and animal eggs. Such proteins act as a reserve source of matter and energy. Some compounds are used by the body as an amino acid reservoir. They, in turn, are precursors of active substances involved in the regulation of metabolism.
Cellular receptors
Such proteins can be located either directly in the cytoplasm or embedded in the wall. One part of the connection receives the signal. As a rule, it is a chemical substance, and in some cases a mechanical effect (stretching, for example), light and other stimuli. In the process of exposure of a signal to a certain fragment of the molecule - the polypeptide receptor - its conformational changes begin. They provoke a change in conformation of the rest of the part that transmits the stimulus to other components of the cell. Sending a signal can be done in different ways. Some receptors are capable of catalyzing a chemical reaction, while others act as ion channels that close or open under the influence of a stimulus. Some compounds specifically bind messenger molecules within the cell.
Motor polypeptides
There is a whole class of proteins that provide movement to the body. Motor proteins are involved in muscle contraction, cell movement, and the activity of flagella and cilia. Due to them, directed and active transport. Kinesins and dyneins transport molecules along microtubules using ATP hydrolysis as an energy source. The latter move organelles and other elements towards the centrosome from peripheral cellular areas. Kinesins move in the opposite direction. Dyneins are also responsible for the activity of flagella and cilia.
Proteins, or proteins, in living organisms are formed mainly from the 20 most important natural amino acids as a result of a polycondensation reaction in the presence of enzymes. The molecular weights of proteins vary over a very wide range: from 10,000 to 1,000,000 and above.
The backbone of the protein chain is built from amino acid fragments connected by peptide bonds and is surrounded by various chemical nature deputies. The peptide bond in proteins is stable at 37°C in a neutral environment, but can be hydrolyzed in an acidic or alkaline environment. In the body, protein hydrolysis is carried out under the action of peptidase enzymes and is strictly controlled.
IN natural proteins The length and composition of the chain vary widely, which allows their molecules, even in solution, to take on diverse conformation.
Conformationsprotein macromolecules in solution represent their various spatial forms, arising as a result of rotations of individual molecular fragments around single bonds and stabilized due to intermolecular bonds between separate groups a given macromolecule or molecules of substances found in the surrounding solution.
Mutual conformational transitions are mainly carried out without breaking covalent bonds in the protein macromolecule. When describing the composition and conformation of a protein, the concepts are used primary, secondary, tertiary And quaternary structure.
Primary structure is specific to an individual protein and is determined by the composition and sequence of amino acid residues of its chain. When writing full formulas proteins indicate the order of amino acid residues following each other using their three-letter designations, starting from the N-terminus of the chain. An idea of the primary structure of human myoglobin, which contains only 153 amino acid residues in the molecule, is given by the following abbreviated notation:
The strictly linear arrangement of the polypeptide chain is energetically unfavorable, since it practically eliminates interactions between different radicals of amino acid residues. As a result of precisely such interactions, additional bonds arise that stabilize one or another conformation of the protein chain in space. This occurs through the following interactions: ion-ion interaction; hydrogen bond; hydration of polar groups; disulfide bond; Vander Waals interactions between non-polar substituents; hydrophobic interactions, as a result of which water molecules are pushed out of the zone of interaction of non-polar substituents with each other, as well as donor-acceptor bond between the complexing ion and the ligand groups of the protein (Fig. 21.3).
Protein secondary structure characterizes the shape of a polypeptide chain, which can be helical (a-structure), folded (B -structure) or disordered (Fig. 21.4). Main role in the formation and maintenance of secondary structure

Rice. 21.3. Types of interactions between substituents of amino acid residues of a protein molecule and the aqueous environment
![]() Rice. 21.4. Secondary structure of proteins: A- a-structure (spiral), b- P-structure (folded) is played by hydrogen bonds that arise between the backbone groups of the polypeptide chain.
Rice. 21.4. Secondary structure of proteins: A- a-structure (spiral), b- P-structure (folded) is played by hydrogen bonds that arise between the backbone groups of the polypeptide chain.
The spatial arrangement of the a-structure can be imagined by imagining that the polypeptide chain wraps around a cylinder, and its side radicals are directed outward. The turns of the helix are held together by hydrogen bonds between peptide groups located on adjacent turns of the helix. And although the energy of these bonds is small, their large number leads to a significant energy effect, as a result of which the a-structure is quite stable and rigid.
Folded (3-structure formed from large number parallel elongated polypeptide chains connected by many hydrogen bonds to each other. The side radicals R are located above and below the plane drawn through the resulting folded sheet.
The disordered structure of individual protein fragments is characterized by a lack of spatial order in their arrangement.
Which secondary structure of a protein is realized depends on its amino acid composition, i.e., on the primary structure. Most natural proteins are characterized by the coexistence in one molecule of fragments with a-, p- and disordered structures.
The low strength of hydrogen bonds makes it relatively easy to transform the secondary structure under external influence: changes in temperature, composition or pH of the environment - or under mechanical influence. As a result of the transformation of the secondary structure of the protein, its native, i.e., primary by nature, properties change, and, consequently, its biological and physiological functions.
Protein tertiary structure determines the general location of its polypeptide chain in space. It is believed that in the formation and stabilization of the tertiary structure of the protein molecule decisive role belongs to the interaction of side amino acid substituents, which are brought closer together in space due to the bends of the polypeptide chain. The types of these interactions were shown in Fig. 21.3.
The tertiary structure of a protein molecule arises completely automatically as a result of self-organization of the polypeptide chain in accordance with its primary and secondary structures, as well as with the composition of the surrounding solution. The driving force that folds the polypeptide chain of a protein into a strictly defined three-dimensional formation is the interaction of amino acid radicals with each other and with the molecules of the surrounding solution. At the same time, in aqueous solutions, hydrophobic substituents are pushed into the protein molecule, forming dry zones there ("fat drops"), and hydrophilic substituents are oriented to the side aquatic environment. At some point, an energetically favorable conformation of the molecule for the aqueous environment is achieved, and this conformation of the protein molecule is stabilized. In this case, the entropy of the polypeptide chain decreases, while the entropy of the system as a whole (polypeptide chain + aqueous medium) remains constant or increases. Thus, from the position of the II law of thermodynamics, the stabilization of the tertiary structure of a protein in an aqueous environment is ensured by the tendency of the aqueous environment of the protein molecule to transition to a state with maximum entropy. An idea of the tertiary structure of the molecules of the proteins myoglobin and lysozyme is given in Fig. 21.5. In the figure, the shaded disk in the myoglobin molecule is a heme containing a porphyrin ligand and a complexing cation, Fe 2+. The lysozyme molecule shows S-S disulfide bridges involved in stabilizing the tertiary structure of this protein.

Rice. 21.5. Tertiary structures: myoglobin (a) and lysozyme (b)
The tertiary structure of a protein, compared to its secondary structure, is even more sensitive to external influences. Therefore, the action of weak oxidizing agents, changes in solvents, changes in ionic strength, pH and temperature disrupt the tertiary structure of proteins, and, consequently, their native properties.
Quaternary structure. Large protein molecules with a molecular weight of more than 60,000 are usually aggregates that consist of several polypeptide chains with a relatively small molecular weight. Moreover, each chain, preserving its characteristic primary, secondary and tertiary structure, acts as a subunit of this aggregate, which has more high level spatial organization - quaternary structure. Such a molecule-aggregate represents a single whole and performs a biological function that is not characteristic of individual subunits. For example, the hemoglobin molecule consists of 4 subunits and is characterized by significantly greater lability of the complex with oxygen than its individual subunits, which is manifested in the properties of myoglobin (section 10.4). The quaternary structure of a protein is fixed primarily by hydrogen bonds and van der Waals interactions, and sometimes by disulfide bonds, between the polypeptide chains being joined. The molecular weight of proteins with a quaternary structure can reach several tens of millions. The quaternary structure of proteins is sensitive to external influences and can be disrupted by them.
The shape of protein molecules. Based on the shape of the molecule, native proteins, i.e. those exhibiting biological properties programmed by nature, are divided into fibrillar And globular. Fibrillar protein molecules usually have a B-structure and a fibrous structure; they do not dissolve in water, since there are many hydrophobic radicals on their surface. Fibrillar proteins are protein fibrons; keratin of hair, skin, nails; collagen of tendons and bone tissue; myosin of muscle tissue.
Globular proteins have a cylindrical or spherical shape and a size of 10 -9 -10 -7 m. They are usually soluble in water, since their surface mainly contains polar groups. Dissolving in water, globular proteins form lyophilic colloidal solutions (Section 27.3). Examples of globular proteins: albumin ( egg white), myoglobin, almost all enzymes.
Liquid crystal state. Protein molecules are quite large formations and have a fixed spatial structure, which can be anisotropic as a whole, or individual fragments of the peptide chain can be anisotropic. Therefore, many proteins are characterized by a liquid crystalline state in a certain temperature range (thermotropic liquid crystalline state) or the formation of one or several lyotropic liquid crystalline states with the participation of an aqueous medium at a certain concentration of substances in solution. The formation of a liquid crystalline state or transitions from one liquid crystalline state to another, accompanied by a change in the orientation of individual fragments of a protein molecule or a change in the consistency of movement in the system, do not require large energy expenditures, but can lead to a change in its biological functions. For example, affect the contractile function of muscle fiber myosin, enzymatic activity, transport function of proteins or their protective properties relative to colloidal systems. Thus, under certain conditions, hemoglobin molecules transform into a liquid crystalline state. This leads to a number of pathological disorders, manifested in the loss of elasticity of red blood cells. As a result, they clog the capillaries and oxygen transport is disrupted. The formation of stones in the urinary or biliary systems is associated with a change not only in concentration, but also in condition protective proteins in these systems. Until recently, the ability of proteins and their solutions to transform into a liquid crystalline state was practically not considered in biology, biochemistry and medicine, despite the extreme importance of these properties from the standpoint of the vital activity of any living systems.
Denaturation. The spatial structure of proteins, as already indicated, can be disrupted under the influence of a number of factors: increased temperature, changes in pH and ionic strength of the medium, UV irradiation and x-rays, the presence of substances capable of dehydrating a protein molecule (ethanol, acetone, urea) or interacting with its substituents (oxidizing agents, reducing agents, formaldehyde, phenol) and even with strong mechanical stirring of solutions.
Denaturation is the destruction of the natural (native) conformation of a protein macromolecule under external influence.
During denaturation, the quaternary, tertiary and secondary structures are destroyed, but the primary structure of the protein is preserved. Therefore, denaturation can be reversible (denaturation - renaturation) and irreversible depending on the nature of the protein and the intensity of external influence. Irreversible denaturation usually occurs when exposed to heat (for example, the coagulation of egg albumin when boiling eggs). Denatured globular proteins have a decreased affinity for water, since many hydrophobic radicals appear on the surface of the molecules. Therefore, their solubility decreases and flakes or sediment appear. The main thing is that during denaturation, the biological activity of both globular and fibrillar proteins is lost, which is observed with many methods of their isolation (Section 11.3). To avoid denaturation of the protein and to preserve its native conformation during the isolation process, all operations are carried out under mild conditions at a temperature not exceeding 5°C, avoiding harsh effects of chemical reagents.
Surface properties of proteins. Protein molecules contain different amino acids, which have hydrophobic radicals based on aliphatic and aromatic hydrocarbons, and hydrophilic radicals, including a peptide group. These radicals are distributed throughout the chain, and therefore most proteins are surfactants (Section 26.6). A characteristic feature of protein surfactants is the presence in their molecules of fragments with sharply different hydrophilic-lipophilic balance, which makes them effective stabilizers for lyophobic disperse systems, emulsifiers of fats and cholesterol, and active components of biological membranes.
Due to their surfactant properties, some proteins form lyophilic micelles (Section 27.3) with lipids (including cholesterol and its esters), called lipoproteins. In lipoproteins there are no covalent bonds between protein and lipid molecules, but only intermolecular interactions. External surface lipoprotein micelle consists of hydrophilic fragments of proteins and phospholipid molecules, and its inner part(core) is a hydrophobic environment in which fats, cholesterol and its esters are dissolved (Fig. 21.6). The presence of a hydrophilic outer shell in lipoproteins makes these lipid-rich micelles “soluble” in water and well suited for transporting fats from the small intestine to fat depots and various tissues. The diameter of lipoprotein micelles ranges from 7 to 1000 nm.
Depending on the density, size of micelles and the ratio of protein and lipids in them, lipoproteins are divided into 4 classes (Table 21.2).

Rice. 21.6. Lipoprotein micelle
The role of chylomicrons and very low density lipoproteins is the transport of fats and their hydrolysis under the action of lipoprotein lipase. As fats are broken down, the following transformation occurs:
P-lipoproteins mainly transport cholesterol into cells, and a-lipoproteins remove excess cholesterol from cells.
When studying the lipoprotein composition of blood serum, it was found that more attitude B-lipoproteins/a-lipo-proteins, the greater the risk of abundant cholesterol deposits on inner surface blood vessels, i.e. atherosclerosis. Atherosclerosis contributes to the development of stroke or myocardial infarction by restricting blood flow through narrowed vessels in the brain or heart.
The surface properties of proteins, characterizing their ability for intermolecular interactions, underlie the interaction of an enzyme with a substrate (Section 5.6), an antibody with an antigen, and explain various interactions, called specific complementarity in biology (the “key and lock” theory). In all these cases, there is a strict correspondence between the surface structure and the properties of the interacting particles, which ensure the high efficiency of various types of intermolecular interactions between them (Fig. 21.3). In biology, this is often reflected in a simplified manner using a graphical correspondence of the shapes and sizes of interacting particles (Fig. 21.7).
Information properties of proteins. Protein molecules and their individual fragments are considered as carriers of biological
Rice. 21.7. Graphical interpretation of the correspondence of intermolecular interactions between protein particles described by specific complementarity or the "key and lock" theory
information in which the role of letters of the alphabet is played by 20 amino acid residues. The reading of this information is based on various types of intermolecular interactions and the system’s desire to use them effectively. For example, in enzymes near the active center, part of the protein molecule contains certain amino acid residues, the substituents of which are oriented in space so that recognition occurs of a strictly defined substrate with which this enzyme reacts. The interaction proceeds similarly antibody- antigen or the synthesis of the corresponding antibody to the emerging antigen occurs in the body. The informational properties of proteins underlie immunity, which is an integral system biological mechanisms self-defense of the body, which are based on information processes of recognizing “friend” and “alien”. “Amino acid language”, containing 20 units, is one of the most optimal and reliable ways of encoding important information for the functioning of living systems, including information about the shape of individual organs and the organism as a whole.
Acid-base properties. Proteins, like a-amino acids (Section 8.2), are polyampholytes, exhibiting acidic properties due to non-ionized carboxyl groups -COOH, ammonium groups of thiol groups -SH, as well as n-hydroxy-
phenyl groups Proteins exhibit their main properties due to the groups - COO-, amino groups - NH 2, as well as imidazole substituents -C 3 H 3 N 2 and guanidine - (CH 5 N 3) +. In aqueous solutions, depending on the pH of the medium, proteins can be present at pH = pI of the protein in a molecular, i.e., neutral form, having a bipolar ionic structure, at pH< рI белка появляется катионная форма, и при рН >pI of the protein appears in an anionic form, mainly due to the ionization of substituents (-RH).

In a strongly acidic environment, the ionized carboxyl group of the protein is protonated, and in a strongly alkaline environment, the terminal ammonium group is deprotonated. However, in biological media, which are not characterized by such extreme pH values, such transformations with protein molecules do not occur. Acid-base transformations in protein molecules are naturally accompanied by a change in their conformation, and therefore, the biological and physiological functions of a protein cation or anion will differ not only from each other, but also from the functions of their molecules.
Depending on the amino acid composition, proteins are divided into “neutral” (pI = 5.0 - 7.0), “acidic” (pI< 4,0) и "основные", или "щелочные" (рI >7.5) (Table 21.3). Acidic proteins have a high content of aspartic or glutamic acids, while “basic” proteins have a high content of arginine, lysine or histidine. Protein buffer systems operate in the body based on proteins (Section 8.4).
The difference in the acid-base properties of proteins underlies the separation and analysis of protein mixtures by electrophoresis and ion exchange chromatography. In a constant electric field, proteins have electrophoretic mobility, and the direction of their movement to the cathode or anode depends on the pH value of the solution and the pI of the protein. At pH< рI белок частично находится в форме катиона и перемещается к катоду. При рН >The pI protein moves to the anode because it is partially in the form of an anion. At pH = pI the protein is completely in molecular form and under the influence electric field doesn't move. The electrophoretic mobility of a protein ion depends on its size and charge, as well as on the pH of the solution. The greater the difference between the pH of the solution and the pH of the protein, the greater the ion mobility. Protein analysis by electrophoresis is widely used in clinical biochemistry for disease diagnosis.
Complexing properties. Proteins are active polydentate ligands (section 10.1), especially containing soft functional groups: thiol, imidazole, guanidine, amino group:

Due to the presence of various functional groups in protein molecules, they form complex compounds of varying stability depending on the polarizability of the complexing ion. With low-polarizable (hard) cations K + and Na +, proteins form low-stable complexes, which in the body act as ionophores for cations or activators of proteins as substrates for certain biochemical processes. With less rigid cations Mg 2+ or Ca 2+, proteins form fairly strong complexes. With cations of d-metals: iron, copper, manganese, zinc, cobalt, molybdenum ("metals of life"), which are sufficiently polarizable, i.e. soft, proteins form strong complexes. However, they form particularly strong complexes with cations of toxic metals: lead, cadmium, mercury and others that exhibit high polarizability, i.e., are very soft. Stable complexes of proteins with metal cations are often called metalloproteins.
Many enzymes are chelate complexes of a protein with a cation of some “metal of life.” In this case, it is the complexing cation that, under the influence of the protein ligand, is active center enzyme, and a fragment of a protein molecule near this center usually serves as an identification and activator of the substrate. The protein component of the metalloenzyme is often called apoenzyme.
All proteins, when treated with copper salts in an alkaline environment, form a violet-colored chelate complex, which is a qualitative reaction to proteins called biuret reaction:
This reaction occurs by deprotonation of the peptide groups of the protein, which is facilitated by the alkaline environment and the presence of a complexing ion in it.
Electrophilic-nucleophilic reactions. These reactions primarily include the hydrolysis of proteins - the main route of their catabolism (breakdown) in the body. During protein hydrolysis, the reagent - a water molecule - acts both as a nucleophile due to OH" and as an electrophile due to H +. The nucleophilic particle OH" attacks the electrophilic center of the peptide bond, i.e., the carbon atom of the carbonyl group, and the nucleophilic center of this bond - nitrogen atom - attacked by an electrophile - a proton. As a result of attack by water molecules, peptide bonds in proteins are broken, and osamino acids and peptides are first formed, and the end products are os-amino acids.
The hydrolytic breakdown of proteins occurs in any cell of the body, more precisely, in its liposomes, where hydrolytic enzymes are concentrated. Protein hydrolysis can be partial (to peptides) and complete (to amino acids). Partial hydrolysis accelerates proteinases, which promote the formation of peptides. The resulting peptides are hydrolyzed to amino acids with the participation peptidase. In the body, protein hydrolysis is carried out mainly by a whole set of enzymes, each of which breaks down the peptide bond formed by certain amino acids. So, carboxypeptidase specifically cleaves the C-terminal amino acid from proteins, trypsin hydrolyzes the peptide bond between amino acids with a non-polar (hydrophobic) substituent. Chymotrypsin cleaves the peptide bond formed by phenylalanine, tyrosine, tryptophan with other amino acids. In the body, food proteins are completely broken down, since mainly free amino acids are used for life.
In laboratory conditions, proteins are hydrolyzed in both acidic and alkaline environments. However, alkaline hydrolysis is practically not used due to the instability of many osaminic acids under these conditions. Typically, complete hydrolysis is carried out by heating the protein to 110°C in a sealed ampoule with 20% HC1 for 24 hours. Under these conditions, protein hydrolysis proceeds to completion, but the resulting tryptophan is completely decomposed. Therefore, preference is given to enzymatic hydrolysis.
Body proteins containing aspartic and glutamic acids can act as an acceptor of ammonia, which, as a nucleophile, reacts at the free carboxyl groups of the substituent, i.e. protein amidation reaction:
The amidation reaction is endergonic, therefore in the body it is associated with the ATP hydrolysis reaction.
For the purpose of sterilizing objects ( complete liberation from microorganisms) they are treated formaldehyde. Formaldehyde, as an active electrophile, reacts at the free amino groups of proteins, forming their methylol derivatives:
As a result of this reaction, the protein loses its native properties, as it is irreversibly denatured.
Active electrophilic reagents (EX): 2,4-dinitrofluorobenzene, phenyl isothiocyanate or dansyl chloride - used to determine the primary structure of proteins or peptides. In the presence of bases, they react at the N-terminal amino acid of the protein anion and promote its elimination in the form of the corresponding derivative E-NH-CRH-COOH, easily identified either chromatographically or spectrally:
The remaining part of the protein is not destroyed, and the operations of removing the next amino acid can be repeated. These reactions underlie the operation of an automatic protein primary structure analyzer. Typically, the protein to be analyzed is first subjected to partial hydrolysis to produce several peptides. The resulting peptides are separated, purified, and the amino acid sequence of each is determined, and then the primary structure of the protein being analyzed is compiled.
Redox properties. Proteins are relatively resistant to mild oxidation, with the exception of those containing the amino acid cysteine, since the thiol group of the latter is easily oxidized into a disulfide group, and the process can be reversible:
As a result of these transformations, a change in the conformation of the protein and its native properties occurs. Therefore, sulfur-containing proteins are sensitive to free radical oxidation or reduction, which occurs when the body is exposed to radiation or toxic forms of oxygen (Section 9.3.9).
Thiol-disulfide transformations of the keratin protein are the basis of chemical hair perm, since cysteine and cystine are part of its composition. First, the hair is treated with a reducing agent to break the -S-S- bonds of cystine and convert it into cysteine thiol groups. Then the hair is styled into ringlets (curled) and treated with an oxidizing agent. In this case, cystine disulfide bonds are formed, which help the hair maintain its new shape.
With more severe oxidation, the thiol group of proteins is oxidized into a sulfo group almost irreversibly:
Hard oxidation of proteins to CO2, H2O and ammonium salts is used by the body to eliminate unnecessary proteins and replenish its energy resources (16.5 - 17.2 kJ/g).
In the body, proteins containing lysine, proline, phenylalanine and tryptophan residues undergo enzymatic hydroxylation (monooxygenase oxidation) with the participation of oxygen and a reduced form of coenzyme:


As a result of the hydroxylation reaction, the hydrophilic properties of the protein and its ability to form hydrogen bonds are enhanced. This occurs in tropocollagen, in which three chains are combined into a stable superhelix due to hydrogen bonds, in the formation of which hydroxyproline residues also participate.
A similar reaction occurs in the tropocollagen molecule, which leads to an even stronger “cross-linking” of its peptide chains.
Oxidative deamination of proteins under the influence of ninhydrin, accompanied by the formation of a blue color - a characteristic qualitative reaction to proteins - ninhydrin reaction(see section 21.2.4).
To detect proteins containing aromatic and heterocyclic amino acids, it is used xanthoprotein reaction, which, when exposed to concentrated nitric acid, is accompanied by the appearance of a yellow color, which turns orange when adding alkali or ammonia:

It is as a result of the xanthoprotein reaction that a yellow coloration of the skin is observed when it comes into contact with concentrated nitric acid.
Thus, proteins are characterized by: a certain conformation, a liquid crystalline state, surface-active and information properties, as well as all four types of chemical reactions: acid-base, complexing, electrophilic-nucleophilic and redox, which underlie the vital activity of any living systems. The combination of all these properties explains the uniqueness of proteins for the entire living world.