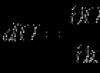Riz. 1. Microscope à épifluorescence
La fluorescence a été décrite pour la première fois en 1852 par le scientifique britannique George Stokes, qui a inventé le terme alors qu'il menait des expériences avec la fluorite (spath fluor), qui émet une lumière rouge lorsqu'elle est irradiée par une lumière ultraviolette. Stokes a remarqué que la longueur d'onde de l'émission fluorescente est toujours supérieure à la longueur d'onde de la lumière d'excitation. Les premières recherches menées au XIXe siècle ont montré que de nombreux échantillons (notamment des minéraux, des cristaux, des résines, des matières premières médicinales, des huiles, de la chlorophylle, des vitamines et des composés inorganiques) émettent une fluorescence lorsqu'ils sont irradiés par une lumière ultraviolette. Cependant, l’utilisation des fluorochromes dans la recherche biologique pour colorer les composants tissulaires, les bactéries et autres agents pathogènes n’a commencé que dans les années 1930. Certains de ces colorants étaient extrêmement spécifiques et ont stimulé le développement de la microscopie à fluorescence.En raison de plusieurs fonctionnalités difficiles à réaliser avec la microscopie optique traditionnelle à contraste amélioré, la microscopie à fluorescence est devenue un outil important dans la recherche biologique et biomédicale et dans la science des matériaux. L'utilisation d'ensembles de fluorochromes a permis d'isoler des cellules très spécifiques et des composants cellulaires submicroscopiques de substances non fluorescentes. Avec un microscope à fluorescence, même des molécules individuelles peuvent être détectées. Grâce à la multicoloration fluorescente, différents colorants peuvent identifier plusieurs molécules cibles simultanément. Bien que la résolution spatiale d'un microscope à fluorescence soit limitée en dessous par la limite de diffraction, qui dépend des caractéristiques spécifiques de l'échantillon, la détection de molécules fluorescentes en dessous de cette limite est tout à fait possible.
De nombreux échantillons présentent une autofluorescence lorsqu’ils sont irradiés (sans utilisation de fluorochromes), et ce phénomène est largement utilisé en botanique, en pétrologie et dans l’industrie des semi-conducteurs. En revanche, l’étude des tissus d’animaux ou d’agents pathogènes est souvent compliquée par une autofluorescence non spécifique extrêmement faible ou, à l’inverse, forte. Beaucoup plus important dans ce cas, il s'agit de l'introduction de fluorochromes (ou fluorophores) dans le tissu, excités à une certaine longueur d'onde et émettant de la lumière avec l'intensité requise. Les fluorochromes sont des colorants qui, se fixant indépendamment sur des structures visibles ou invisibles, ont une sélectivité élevée envers les cibles et un rendement quantique élevé (le rapport entre le nombre de photons émis et le nombre de photons absorbés). La croissance rapide de l’utilisation de la microscopie à fluorescence est étroitement liée à l’émergence de nouveaux fluorophores synthétiques et naturels qui présentent des profils d’intensité d’excitation et d’émission spécifiques et sont « ciblés » sur des cibles biologiques données.
Bases des processus d'excitation et d'émission
Le principe de fonctionnement d'un microscope à fluorescence consiste à irradier un échantillon à des longueurs d'onde dans la plage requise et définie avec précision, suivi de la séparation de la fluorescence émise beaucoup plus faible du flux de lumière excitatrice. Dans un microscope bien réglé, seule la lumière émise doit atteindre l’œil ou l’appareil récepteur, de sorte que les structures fluorescentes observées se superposent sur un fond très contrasté et très sombre (ou noir). L’obscurité de l’arrière-plan détermine généralement les limites de détection, puisque la lumière d’excitation est généralement des centaines de milliers, voire des millions de fois plus brillante que la fluorescence émise.La figure 1 montre une coupe schématique d'un microscope à épifluorescence moderne, conçu pour les observations en lumière transmise et réfléchie. L'éclairage vertical, situé au centre, comporte une source lumineuse à une extrémité (indiquée dans le schéma comme un module épiscopique) et un support de filtre à l'autre. La conception est basée sur un microscope fonctionnant en lumière réfléchie dont la longueur d’onde est supérieure à la longueur d’onde d’excitation. L'auteur de l'illuminateur vertical pour la microscopie à fluorescence en lumière réfléchie est considéré comme étant Johan S. Ploem. La lumière multifréquence provenant d'une lampe à arc ou d'une autre source, passant à travers un filtre d'excitation sélectif, est convertie dans un éclairage vertical fluorescent en lumière avec une certaine longueur d'onde (ou dans un intervalle d'onde donné), généralement à partir des parties ultraviolettes, bleues ou vertes. du spectre. Le flux traversant le filtre d'excitation est réfléchi par la surface d'un miroir dichromatique (également appelé dichroïque) ou d'un séparateur de faisceau et, en passant à travers l'objectif, illumine l'échantillon avec une lumière intense. Si l'échantillon est fluorescent, la lumière émise, collectée par l'objectif, traverse à nouveau le miroir dichroïque et est ensuite filtrée par un filtre de blocage (ou d'émission), qui bloque la lumière aux longueurs d'onde d'excitation. Il est important de noter que la fluorescence est le seul mode en microscopie optique dans lequel l’échantillon émet sa propre lumière après irradiation. La lumière émise est réémise de manière sphérique dans toutes les directions, quel que soit l'emplacement de la source lumineuse irradiante.
L'éclairage épifluorescent est la méthode prédominante dans microscopie moderne. L'illuminateur vertical à lumière réfléchie est situé entre les tubes d'observation et la tête tournante des lentilles. L'illuminateur est conçu de telle manière que la lumière d'excitation en direction de l'image et en provenance de l'échantillon traverse le même objectif du microscope, qui dans cette configuration agit d'abord comme un condenseur, et collecte sur le chemin de retour la lumière de fluorescence émise. Les briquets de ce type présentent plusieurs avantages. La lentille d'un microscope à fluorescence agit, d'une part, comme un condenseur bien réglé, et, d'autre part, comme un dispositif de collecte de lumière avec lequel une image est formée. Étant le même composant, l’objectif/condensateur est toujours parfaitement aligné. La majeure partie de la lumière d'excitation atteignant l'échantillon le traverse sans interaction et ne retourne pas vers l'objectif, et la zone éclairée est limitée à la partie de l'échantillon observée à travers les oculaires (dans la plupart des cas). Si le microscope est correctement configuré pour l'éclairage de Köller, contrairement à certaines techniques d'amélioration du contraste, la pleine ouverture numérique de l'objectif est disponible pour la visualisation. De plus, il permet de combiner les modes d'observation en lumière transmise et réfléchie, le mode de formation d'image numérique, ou d'en sélectionner un.

Riz. 2. Filtres fluorescents
Comme le montre la figure 1, à l'extrémité arrière de l'illuminateur à lumière réfléchie verticale se trouve une lampe à arc (généralement au mercure ou au xénon). Se propageant le long de l'illuminateur perpendiculairement à l'axe optique du microscope, la lumière d'excitation traverse les lentilles collectrices, le diaphragme à ouverture réglable et centrable, puis à travers le diaphragme de champ réglable et centrable (voir Figure 1). Après cela, la lumière pénètre dans le filtre d'excitation, où les longueurs d'onde sont sélectionnées dans l'intervalle requis et les longueurs d'onde restantes sont bloquées. Après avoir traversé le filtre d'excitation, les longueurs d'onde sélectionnées atteignent le miroir de séparation de faisceau dichroïque, qui est un filtre d'interférence spécial qui reflète efficacement la lumière de courte longueur d'onde et transmet efficacement la lumière de longue longueur d'onde. Le séparateur de faisceau dichroïque est incliné à un angle de 45 degrés par rapport à la lumière d'excitation incidente et la réfléchit à un angle de 90 degrés à travers la lentille du système optique directement sur l'échantillon. La fluorescence émise par l'échantillon éclairé est captée par la lentille, qui remplit désormais sa fonction habituelle, à savoir la formation d'images. Étant donné que les longueurs d'onde émises sont plus longues que les longueurs d'onde d'excitation, elles traversent le miroir dichroïque vers le haut jusqu'aux tubes d'observation ou au détecteur d'électrons.La majeure partie de la lumière d’excitation diffusée atteignant le miroir dichroïque est réfléchie vers la source lumineuse, bien qu’une petite fraction puisse traverser ou être absorbée par le revêtement interne du miroir. Mais avant que la fluorescence émise n'atteigne l'oculaire ou le détecteur, elle doit traverser un filtre de blocage ou de blocage. Ces filtres bloquent (bloquent) toute lumière d'excitation résiduelle, mais en autorisent davantage. longues vagues lumière émise. La plupart des illuminateurs à lumière réfléchie combinent le filtre d'excitation, le miroir dichroïque et le filtre d'arrêt dans une unité optique (souvent appelée cube), comme le montre la figure 2. Les microscopes à fluorescence modernes peuvent accueillir quatre à six cubes filtrants (généralement sur un type carrousel ou tiroir). (accessoire) ; voir Figure 1) et permettent à l'utilisateur d'installer facilement des filtres d'excitation, des filtres de blocage et des miroirs dichroïques remplaçables.
La conception de l'illuminateur vertical doit permettre à l'utilisateur d'ajuster le microscope pour l'éclairage de Köller, qui fournit un éclairage brillant et uniforme sur tout le champ de vision. Les lentilles à condensateur corrigées du système optique garantissent que l'image du diaphragme à ouverture centrée est alignée avec l'ouverture arrière de l'objectif de mise au point. Dans les éclairages modernes, l'image d'un diaphragme à champ centré et préfocalisé est conjuguée à l'image focalisée et au plan du diaphragme fixe de l'oculaire.
L'unité de lampe éclairante contient généralement un filtre d'arrêt qui bloque la lumière infrarouge. La lampe elle-même ne doit pas transmettre de rayonnement ultraviolet vers l’extérieur. Il est également souhaitable qu'il dispose d'un disjoncteur intégré au cas où il s'ouvrirait pendant le fonctionnement. La lampe doit être suffisamment solide pour résister à une éventuelle explosion de la lampe à arc pendant le fonctionnement. Dans les lampes modernes, la douille de la lampe est équipée de boutons de réglage pour centrer l'image de la lampe à arc dans l'ouverture arrière de l'objectif (sous éclairage Keller, ces plans sont conjugués). Il est conseillé de placer un obturateur sur le trajet de la lumière, généralement à proximité de la lampe mais avant le filtre d'excitation, pour bloquer complètement la lumière d'excitation à moins que l'échantillon ne soit observé. De plus, l'équipement de l'illuminateur doit comprendre des filtres à densité neutre (sur fixation de type tambour, carrousel ou rétractable) afin de pouvoir réduire l'intensité de l'éclairage d'excitation.
Changement de Stokes
Lorsque les électrons passent de l’état excité à l’état fondamental, ils perdent énergie vibratoire. En raison de cette perte d'énergie, le spectre d'émission du fluorophore excité se déplace généralement vers des longueurs d'onde plus longues par rapport au spectre d'absorption ou d'excitation (rappelez-vous que la longueur d'onde est inversement proportionnelle à son énergie). Ce phénomène bien connu est appelé règle de Stokes ou décalage de Stokes. À mesure que le décalage de Stokes augmente, il devient plus facile de séparer la lumière d’excitation et d’émission à l’aide de combinaisons de filtres fluorescents.L'intensité maximale d'émission d'un fluorophore est généralement inférieure à l'intensité maximale de son absorption et se produit à une longueur d'onde plus longue. La courbe d'émission (courbe spectrale) est souvent une image miroir (ou proche) de la courbe d'excitation, mais décalée vers des longueurs d'onde plus longues, comme le montre la figure 3, qui montre les caractéristiques spectrales utiles du colorant Alexa Fluor 555, qui absorbe dans le jaune-vert et émet dans la région jaune-orange. Pour obtenir une intensité de fluorescence maximale, le fluorophore (souvent appelé colorant) est excité à des longueurs d'onde proches ou proches du pic de la courbe d'excitation, et la lumière émise est détectée sur la plage la plus large possible incluant le pic d'émission. La sélection des longueurs d'onde excitatrices et émises est réalisée à l'aide de filtres interférentiels (Figure 2). De plus, il convient de noter que les caractéristiques spectrales du système optique du microscope dépendent également de la transmission du verre (qui est affectée par les revêtements antireflet), du nombre de lentilles et de miroirs et de la sensibilité des détecteurs.

Riz. 3. Courbes d'absorption et d'émission des fluorophores
L'efficacité de la séparation et de l'enregistrement des longueurs d'onde d'excitation et d'émission est obtenue en microscopie à fluorescence le bon choix des filtres de lumière qui bloquent ou, à l'inverse, transmettent la lumière de certaines longueurs d'onde dans les régions ultraviolettes, visibles et proches infrarouges du spectre. La lumière d'excitation est contrôlée dans les éclairages fluorescents verticaux du fait que leur conception prévoit l'utilisation de filtres facilement remplaçables (filtres d'excitation neutres et interférentiels) insérés le long du trajet lumineux vers l'échantillon et sur le trajet de retour entre l'échantillon et les tubes d'observation. ou système de réception de signal. En raison de la faible intensité de l'émission fluorescente (comme indiqué ci-dessus), il est nécessaire que la source de lumière d'excitation ait une luminosité suffisante pour maximiser la faible lumière émise, et que les fluorochromes aient des caractéristiques d'absorption et une efficacité quantique appropriées. C’est peut-être le critère clé de la microscopie à fluorescence.
L'efficacité avec laquelle un fluorophore individuel absorbe un photon de lumière d'excitation est fonction de sa section efficace moléculaire, et la probabilité qu'un tel événement se produise est appelée coefficient d'absorption. Des valeurs plus grandes du coefficient d'absorption indiquent que l'absorption d'un photon (ou d'un quantum) dans un intervalle de longueur d'onde donné est plus probable. Le rendement quantique est le rapport entre le nombre de quanta émis et le nombre de quanta absorbés (il se situe généralement entre 0,1 et 1,0). Le fait que le rendement quantique prenne des valeurs inférieures à 1 est une conséquence d'une perte d'énergie de manière non radiative, par exemple par la chaleur ou une réaction photochimique, lorsque la réémission ne se produit pas, conduisant à la fluorescence. Le coefficient d'absorption, l'efficacité quantique, l'intensité lumineuse moyenne et le temps de luminescence sont facteurs importants, affectant l'intensité de la fluorescence et déterminant la faisabilité de l'utilisation de cette méthode.
Décoloration, décoloration et photoblanchiment
Un certain nombre de conditions peuvent affecter la probabilité de réémission de fluorescence, entraînant souvent une baisse de l'intensité de la fluorescence. Terme général pour indiquer une diminution de l'intensité de l'émission fluorescente s'estompe, couvrant tous les phénomènes qui sont plus description détaillée peut être divisé en phénomènes de trempe et de photoblanchiment. Le photoblanchiment est la dégradation irréversible de molécules fluorescentes dans un état excité, provoquée par leur interaction avec l'oxygène moléculaire avant leur émission. Ce phénomène est utilisé dans la méthode de récupération de fluorescence après photoblanchiment (FRAP), très efficace pour étudier les propriétés de diffusion et le mouvement des macromolécules biologiques. La méthode est basée sur le photoblanchiment avec un faisceau laser d’une zone clairement définie de l’échantillon, suivi de l’observation du taux et de la nature de la récupération de fluorescence dans la zone photoblanchie. Une technique connexe, la décroissance de la fluorescence dans les images blanchies (FLIP), est utilisée pour étudier la diminution de la fluorescence dans les régions adjacentes à la région photoblanchie. Comme FRAP, cette méthode est un outil efficace pour étudier la mobilité et la dynamique moléculaire des cellules vivantes.
Riz. 4. Taux de photoblanchiment d’échantillons multicolores
La figure 4 montre un exemple typique de photoblanchiment (décoloration) observé dans la série images numériques culture multicolorée de fibroblastes cutanés de muntjac indien prélevés divers moments temps. Les noyaux ont été colorés avec un dérivé de bis-benzimidazole (Hoechst 33258, fluorescence bleue), et les mitochondries et le cytosquelette d'actine ont été colorés avec MitoTracker Red CMXRos (fluorescence rouge) et un dérivé de phalloïdine couplé à Alexa Fluor 488 (fluorescence verte), respectivement. Des images ont été prises toutes les deux minutes et la combinaison de filtres fluorescents a été ajustée de manière à ce que les trois fluorophores soient excités simultanément, tout en enregistrant simultanément les signaux émis combinés. La figure 4 (a) montre que l'intensité des trois fluorophores est relativement élevée, mais que l'intensité de Hoechst (bleu) commence à diminuer rapidement après seulement deux minutes et disparaît presque complètement au bout de 6 à 8 minutes. Les colorants mitochondriaux et actines semblent plus résistants au photoblanchiment, mais leur intensité diminue également de manière significative au cours de la période d'observation (10 minutes).La relaxation de l'état excité par trempe, entraînant une baisse de l'intensité de la fluorescence, se produit de diverses manières non radiatives et est souvent due à des agents oxydants ou à la présence de sels, de métaux lourds et de composés halogènes. Dans certains cas, l'extinction se produit à la suite du transfert d'énergie vers une autre molécule (appelée accepteur) proche du fluorophore excité (donneur). Ce phénomène est appelé transfert d'énergie par résonance de fluorescence (FRET). C'est ce mécanisme qui est devenu la base méthode efficaceétudier interactions moléculaires et des associations à des distances nettement inférieures à la résolution des microscopes optiques.
Sources lumineuses fluorescentes
Une conséquence malheureuse de la faible intensité d’émission dans la plupart des applications de microscopie à fluorescence est le faible nombre de photons atteignant l’oculaire ou le récepteur. Dans la plupart des cas, l'efficacité de la collecte de photons dans les microscopes optiques est inférieure à 30 pour cent et les concentrations de nombreux fluorophores le long du trajet optique vont de concentrations micromolaires à nanomolaires. Pour que l’intensité lumineuse d’excitation soit suffisante pour détecter la fluorescence, de puissantes sources lumineuses compactes, telles que de petites lampes à arc à haute énergie, sont nécessaires. Les plus courantes sont les lampes au mercure d'une puissance de 50 à 200 watts et les lampes au xénon d'une puissance de 75 à 150 watts (voir figure 5). Ces lampes sont généralement alimentées par une source externe CC, de quoi s'enflammer décharge en arc grâce à l'ionisation de la vapeur à haute pression et maintenir sa combustion avec un scintillement minimal.L'alimentation de la lampe à arc externe du microscope est généralement équipée d'une minuterie pour suivre le nombre d'heures d'utilisation. Les lampes à arc perdent leur puissance lumineuse et tombent souvent en panne lorsqu'elles sont utilisées au-delà de leur durée de vie spécifiée (200 à 300 heures). Les lampes au mercure ne fournissent pas une intensité uniforme sur toute la plage spectrale allant des UV aux IR. Leur intensité maximale se produit dans le proche ultraviolet. Des pics d'intensité distincts se produisent à 313, 334, 365, 406, 435, 546 et 578 nanomètres. Aux autres longueurs d’onde du spectre visible, l’intensité est stable, bien que moins élevée (mais néanmoins suffisante pour la plupart des applications). Mais la puissance de la lampe elle-même n’est pas un paramètre déterminant pour l’efficacité de l’éclairage. A l'inverse, un paramètre essentiel qu'il faut en premier prendre en compte est la luminosité moyenne, prenant en compte la luminosité de la source, la géométrie de l'arc et la répartition angulaire du rayonnement.

Riz. 5. Lampes fluorescentes à arc
Ces dernières années, la microscopie optique a connu un regain d'applications. source laser lumière, en particulier les lasers à ions argon et argon-krypton (ions). Les avantages de ces lasers sont leur petite taille, leur faible divergence de faisceau, leur degré élevé de monochromaticité et leur luminosité moyenne élevée. Ils sont largement utilisés en microscopie confocale à balayage, devenue outil puissant créer des images fluorescentes à contraste élevé en éliminant l'éclairage flou provenant du plan focal de l'échantillon. Dans les microscopes confocaux, ceci est réalisé en balayant l'échantillon avec un point focal ou une ligne tout en formant simultanément une image à travers une ouverture conjuguée. Les sections optiques des échantillons peuvent être stockées dans la mémoire de l'ordinateur du microscope et reconstruites en une image finale affichée sur un moniteur.Symboles de filtre
La terminologie générale adoptée pour désigner les combinaisons de filtres en microscopie à fluorescence est devenue assez confuse en raison des diverses abréviations et codes utilisés. par différents fabricants pour marquer vos filtres. En principe, il existe trois grandes catégories de filtres : les filtres d'excitation (souvent simplement appelés excitateurs), les filtres de blocage (d'émission) et les séparateurs de faisceaux dichroïques (ou miroirs dichroïques). Auparavant, les filtres fluorescents étaient constitués uniquement de verre coloré ou de gélatine pris en sandwich entre deux plaques de verre. Cependant, il existe aujourd'hui une tendance à produire des filtres très sensibles avec une optique interférentielle pour transmettre ou retarder la lumière de longueurs d'onde strictement définies, qui ont également une transmission élevée. Les séparateurs de faisceau dichroïque sont des filtres interférentiels spéciaux conçus pour réfléchir ou transmettre la lumière de longueurs d'onde spécifiques lorsqu'ils sont placés à un angle de 45 degrés le long du trajet lumineux (voir figures 1 et 2). Les filtres bloquants sont fabriqués à partir de verre coloré ou de revêtements interférentiels (ou une combinaison des deux).Les fabricants utilisent différentes abréviations pour indiquer les caractéristiques des filtres d'excitation. Le verre ultraviolet, par exemple, est désigné UG et le verre bleu, BG. Sur les filtres à bande étroite, vous pouvez souvent voir la désignation KP (K de l'allemand « kurz », qui se traduit par « court ») ou simplement SP. Les filtres antiparasitaires sont désormais étiquetés par certains fabricants avec l'abréviation IF. Les filtres d'excitation interférentielle à bande étroite sont particulièrement efficaces pour les faibles déplacements de Stokes.
Les abréviations et abréviations des filtres bloquants sont les suivantes : LP ou L pour les filtres à large bande, Y ou GG pour le verre jaune (de l'allemand "gelb" - jaune), R ou RG pour le verre rouge, OG ou O pour le verre orange, K pour filtres à fentes (de l'allemand « kante » - bord) et BA pour bloquer les filtres. Si un filtre porte un numéro, tel que BA515, il indique la longueur d'onde (en nanomètres) à laquelle il a la moitié de sa transmission maximale.
Les séparateurs de faisceau dichroïque portent également diverses abréviations : CBS signifie séparateur de faisceau chromatique, DM signifie miroir dichroïque, TK signifie séparateur de fentes (de l'allemand "teiler kante"), FT signifie séparateur de couleurs (de l'allemand "farb teiler"). "), et RKP signifie réflecteur à bande étroite. Toutes ces désignations sont interchangeables. De plus, le verre optique de tous les séparateurs de faisceau dichroïque modernes est toujours recouvert de revêtements interférentiels (plutôt que de colorants organiques ou métalliques). une haute réflectivité. ondes courtes et transmission élevée des ondes longues. Les séparateurs de faisceau dichroïque sont inclinés à un angle de 45 degrés par rapport à la lumière d'excitation incidente sur l'ensemble optique à travers l'illuminateur fluorescent à lumière réfléchie. Leur fonction principale est de rediriger certaines ondes excitatrices (plus courtes) vers la lentille et vers l'échantillon situé derrière elle. Ces filtres spéciaux ont également pour fonction supplémentaire de transmettre des longueurs d'onde de fluorescence plus longues au filtre de blocage et de réfléchir la lumière d'excitation diffusée vers la lampe.

Riz. 6. Filtre d'excitation bleu mi-bande Nikon B-2E
La figure 6 montre les courbes de transmission pour une combinaison de filtres fluorescents typiques utilisés dans les microscopes modernes. Le spectre du filtre d'excitation (courbe rouge) présente une transmission élevée (environ 75 %) dans la plage de 450 à 490 nanomètres avec une longueur d'onde centrale (CWL) de 470 nanomètres. Le miroir dichroïque (courbe jaune) réfléchit les ondes dans le domaine spectral du filtre d'excitation, mais transmet, avec un coefficient relativement élevé, des ondes de plus en plus courtes. Il convient de noter que la transmission nulle d'un miroir dichroïque correspond à une réflexion de 100 pour cent. Le creux distinct de la courbe de transmission entre 450 et 500 nanomètres, qui correspond au pic de réflexion, sert à rediriger les ondes de la bande passante du filtre d'excitation à 90 degrés vers l'échantillon. Le dernier maillon de cette séquence est le filtre d'émission ou de blocage (courbe blanche), qui transmet des ondes dans la partie verte du spectre visible dans la plage de 520 à 560 nanomètres. Pour assurer une séparation quasi complète des ondes réfléchies et transmises, les limites des bandes de réflexion et de transmission des différents spectres superposés doivent être aussi raides que possible. La partie sinusoïdale de la courbe spectrale du miroir dichroïque, appelée sonnerie, est le résultat du processus de dépôt de couches minces. La haute efficacité de cette combinaison de filtres est un exemple d’avancées significatives dans la technologie des filtres interférentiels à couche mince.Au cœur du système symboles, utilisés par Nikon, sont des termes mixtes apparus au début des années 1990. À cette époque, toutes les combinaisons de filtres supplémentaires de Nikon étaient produites à l'aide de la méthode de revêtement dur, mais aujourd'hui, de nombreux filtres sont fabriqués à l'aide de méthodes avancées de revêtement doux. Et bien que les revêtements souples soient plus sensibles à l'humidité et à la chaleur et nécessitent une manipulation plus prudente (par rapport aux revêtements durs), ils démontrent davantage valeurs élevées densité optique et offrent une plus grande facilité de réglage précis des bandes passantes spécifiques. Comprendre les conventions des combinaisons de filtres Nikon vous permet de sélectionner rapidement les filtres nécessaires pour des fluorophores spécifiques.
La première lettre du système de notation alphanumérique de Nikon indique la région du spectre d'excitation (par exemple, UV, V, B et G sont les abréviations de Mots anglais"ultraviolet" - ultraviolet, "violet" - violet, "bleu" - bleu et "vert" - vert, respectivement). Le chiffre suivant le codage du spectre d'excitation indique la bande passante du filtre d'excitation : 1 correspond à une excitation bande étroite, 2 à une excitation bande moyenne et 3 à une excitation large bande. Enfin, une ou plusieurs lettres suivant le chiffre correspondant à la bande passante d'excitation indiquent les caractéristiques du filtre bloquant. La lettre A indique le filtre de coupure large bande standard avec la longueur d'onde de coupure la plus basse, B indique le filtre de coupure large bande ayant plus haute vague coupures. La désignation E (de l'anglais « Enhanced » - Enhanced) dans les filtres d'émission passe-bande indique des performances améliorées dans le sens de réduction de l'interaction interférentielle des signaux séparés. La désignation E/C indique une combinaison de revêtements interférentiels doux conçus spécifiquement pour fonctionner avec des colorants spécifiques tels que le DAPI, le FITC, le TRITC et le Texas Red.
Balance de lumière fluorescente
L'évaluation des flux lumineux d'un microscope à fluorescence typique donne une idée générale des limitations qui surgiront inévitablement lors de la génération d'images numériques ou de l'observation visuelle d'échantillons. Comme source d'irradiation pour notre évaluation, nous prendrons une lampe à arc au xénon standard de 75 watts, densité moyenne dont le flux lumineux est d'environ 400 candelas par millimètre carré (d'autres sources sont présentées dans le tableau 1). Lorsque la lumière émise est dirigée vers un filtre interférentiel de 490 nanomètres (avec une bande passante de 10 nanomètres et une transmission de 75 %), environ 2 milliwatts de puissance de la lampe le traversent. Après réflexion sur un miroir dichroïque avec un coefficient de 0,9, un flux lumineux de 1,8 milliwatts est dirigé vers l'ouverture arrière de l'objectif du microscope sous forme de faisceau d'excitation.Pour un objectif 100x avec une ouverture numérique de 1,4, la zone éclairée de l'échantillon sera de 12 x 10 E(-6) centimètres carrés, si le diamètre du champ de vision est supposé être d'environ 40 micromètres. Le flux lumineux incident sur l’échantillon sera alors d’environ 150 watts par centimètre carré, ce qui correspond à une densité de flux de 3,6 × 10 E (20) photons par centimètre carré. Ainsi, l’intensité d’éclairage de l’échantillon est environ 1000 fois supérieure à l’intensité d’éclairage surface de la terre par une journée ensoleillée normale.
L'émission fluorescente à un tel flux lumineux dépend des propriétés d'absorption et d'émission du fluorophore, de sa concentration dans l'échantillon et de la longueur du trajet optique de l'échantillon. La fluorescence (F) produite mathématiquement est décrite par l'équation :
F = σ Q je
Où σ est la section efficace d'absorption moléculaire, Q est le rendement quantique et I est le flux lumineux incident calculé ci-dessus. En supposant que la fluorescéine est un fluorophore avec une section efficace d’absorption (σ) de 3 × 10 E(-16) centimètres carrés, nous obtenons un Q de 0,99, ce qui se traduit par une fluorescence F de 100 000 photons par seconde et par molécule. À une concentration de colorant de 1 micromole par litre, uniformément répartie dans un disque de 40 micromètres de diamètre et 10 micromètres d'épaisseur (volume égal à 12 picolitres), cela donne environ 1,2 x 10 E(-17) moles de colorant, soit 7,2 millions molécules dans le chemin optique. Si toutes les molécules sont excitées simultanément, le taux de fluorescence sera de 7,2 x 10 photons E(11) par seconde (qui est le produit de F et du nombre de molécules de colorant). La question se pose : combien de photons émis seront enregistrés, et combien de temps ce taux d’émission peut-il perdurer ?
Tableau 1. Densité d'énergie lumineuse de diverses sources lumineuses
L'efficacité de la détection des photons est déterminée par l'efficacité de leur collecte et le rendement quantique du détecteur. Un objectif avec une ouverture numérique de 1,4 et une transmission de 100 pour cent (ce qui est une condition irréaliste) a une efficacité maximale de collecte de photons limitée à un angle d'acceptation d'environ 30 pour cent. La transmission du miroir dichroïque est de 85 pour cent et celle du filtre bloquant est de 80 pour cent. L’efficacité de collecte qui en résulte dans ce cas est de 20 %, soit 140 milliards de photons par seconde. Si un dispositif à couplage de charge (CCD) traditionnel est utilisé comme détecteur, son rendement quantique à la longueur d'onde de la fluorescéine verte (525 nanomètres) sera de 50 %. Ainsi, 70 milliards de photons par seconde seront détectés, soit environ 10 % de ceux émis par fluorescence. Même un détecteur idéal (avec une efficacité quantique de 100 %) ne peut capturer qu’environ 20 % des photons de fluorescence.
La durée de la lueur fluorescente dépend du taux de destruction des fluorophores, conséquence du photoblanchiment. Les mesures montrent que chaque molécule de fluorescéine présente dans une solution saline contenant de l'oxygène parvient à émettre environ 36 000 photons avant d'être détruite. Dans un environnement sans oxygène, le taux de photodestruction est réduit d’environ dix fois. Ainsi, une molécule de fluorescéine peut produire 360 000 photons. Collectivement, tous les colorants de notre exemple (7,2 millions de molécules) sont capables d’émettre un minimum de 2,6 x 10 E(11) et un maximum de 2,6 x 10 E(12) photons. A un taux d'émission d'une molécule de 100 000 photons par seconde (d'après les estimations ci-dessus), on obtient la durée d'émission fluorescente avant photodestruction égale à 0,3 à 3 secondes. Si 10 pour cent des photons émis sont détectés, le signal du détecteur sera de 7,2 × 10 E(10) électrons par seconde.
Ainsi, si un CCD dispose d'une caméra de 1 000 x 1 000 pixels, ce signal sera réparti entre un million d'éléments photosensibles, soit environ 72 000 électrons pour chacun d'eux. Pour un CCD de recherche doté d'éléments photosensibles de 9 micromètres, la capacité de charge est d'environ 80 000 électrons et le bruit de lecture est inférieur à 10 électrons. Dans ce cas, le rapport signal sur bruit sera principalement déterminé par le bruit de fluctuation des photons égal à racine carrée signal, soit environ 268. Dans presque tous les cas, ce haut niveau Le signal ne peut durer que peu de temps avant que la photodestruction ne se produise. Pour prolonger la durée d'observation, la plupart des microscopistes réduisent l'intensité du flux irradiant de sorte que seule une partie du nombre total de molécules fluorophores soit excitée, et donc détruite. Ainsi, le rapport signal sur bruit atteint rarement le maximum théorique et se situe généralement entre 10 et 20 en microscopie à fluorescence.
Détection d'une seule molécule
Dans des conditions idéales, il est souvent possible de détecter l’émission fluorescente d’une seule molécule, à condition, bien entendu, que le fond optique et le bruit du détecteur soient suffisamment faibles. Comme mentionné ci-dessus, une molécule de fluorescéine peut émettre jusqu'à 300 000 photons avant d'être détruite par photoblanchiment. Avec une efficacité de collecte et de détection de 20 %, environ 60 000 photons seront détectés. En utilisant des CCD basés sur des photodiodes à avalanche ou sur la multiplication d'électrons pour des expériences de ce type, les chercheurs ont pu surveiller le comportement de molécules individuelles sur une période de quelques secondes, voire minutes. Le problème principal Dans de tels cas, le bruit de fond optique est supprimé. Étant donné que de nombreux matériaux utilisés dans la construction de lentilles et de filtres microscopiques présentent un certain degré d’autofluorescence, les efforts initiaux ont été orientés vers la production de composants à faible fluorescence. Cependant, il est vite devenu évident qu'en utilisant la méthode de microscopie complète à fluorescence réflexion interne(TIR, ou TIR dans son acronyme anglais), la combinaison requise de lumière de fond faible et de lumière d'excitation de haute intensité peut être obtenue.

Riz. 7. Configurations de microscopes inversés et TIRF
La microscopie à fluorescence par réflexion interne totale (TIRFM ou TIRFM dans son acronyme anglais) utilise le phénomène d'onde non propagée ou décroissante rapidement qui se produit lors de la réflexion interne totale à l'interface de deux milieux d'indices de réfraction différents.
Un circuit utilisant une source de lumière externe est illustré à la figure 7 (a). Dans cette méthode, un faisceau de lumière (généralement un faisceau laser étendu) traverse un prisme à indice de réfraction élevé (comme du verre ou du saphir) adjacent au verre ou à une solution aqueuse à indice de réfraction inférieur. Si la lumière est dirigée vers un prisme selon un angle supérieur à l’angle critique, le faisceau sera complètement réfléchi par l’interface. Le phénomène de réflexion provoque une onde non propagée à l'interface, c'est-à-dire qu'un champ électromagnétique est généré qui pénètre dans un milieu avec un indice de réfraction inférieur jusqu'à une distance ne dépassant pas 200 nanomètres. L'intensité lumineuse de l'onde évanescente est suffisante pour exciter les fluorophores, mais en raison de sa profondeur extrêmement faible, la quantité d'excitation est très faible. Il en résulte un bruit de fond de faible niveau, car le volume de l'échantillon exposé à l'irradiation est négligeable (uniquement la partie située à moins de 200 nm de la surface).
La microscopie à fluorescence par réflexion interne totale peut également être réalisée à l'aide d'une technique d'épi-illumination modifiée utilisée en microscopie à grand champ (comme le montre la figure 7 (b)). Cette méthode nécessite des objectifs à NA très élevés (au moins 1,4, mais de préférence 1,45 à 1,6) et un champ de microscope partiellement éclairé, obtenu en utilisant un petit point ou, pour une plus grande uniformité d'éclairage, un anneau fin qui bloque une partie de la lumière. flux. Pour atteindre l'angle critique au-delà duquel la réflexion interne totale se produit, il est nécessaire que le milieu d'immersion dans les lentilles et le verre de protection du microscope ait un indice de réfraction élevé. Comme le montre la figure 7(b), rayons lumineux, émergeant de la lentille frontale sous un angle inférieur à l'angle critique (sur la figure il est désigné A(1)), ne reviennent plus au microscope. Lorsque l'angle critique est atteint ou dépassé (angle A(2) sur la figure 7(b)), une réflexion interne totale se produit.
Pour fournir des informations supplémentaires dans la recherche, la réflexion interne totale est souvent combinée à d'autres techniques avancées de fluorescence populaires telles que le transfert d'énergie par résonance de fluorescence (FRET), la récupération de fluorescence après photoblanchiment (FRAP) et la spectroscopie. En combinaison, ces méthodes constituent des outils puissants dans l’étude de fluorophores individuels et de molécules fluorescentes. Les avantages de l’étude de molécules uniques commencent seulement à se faire sentir. Ainsi, aujourd’hui, l’éventail des études en microscopie optique s’étend des molécules uniques aux animaux entiers.
Conclusion
Les microscopes à fluorescence modernes combinent la puissance de composants optiques de haute qualité avec un contrôle informatisé et une imagerie numérique pour atteindre un niveau de sophistication qui dépasse de loin la simple observation visuelle. Aujourd'hui, la microscopie s'appuie fortement sur les techniques d'imagerie électronique pour obtenir rapidement des informations à partir de niveaux bas signaux lumineux ou à des longueurs d'onde visuellement non détectables. Ces améliorations techniques ne sont pas seulement des éléments de conception externe, mais aussi des composants essentiels du microscope optique en tant que système de mesure complexe.
Le temps où la microscopie optique était une discipline purement descriptive ou un jouet intellectuel est révolu. Aujourd’hui, l’imagerie optique n’est que la première étape de l’analyse des données. Cette première étape est réalisée au microscope en collaboration avec détecteurs électroniques, processeurs d'images, écrans, qui peuvent être considérés comme une extension du système d'imagerie. Le contrôle informatisé de la mise au point, de la position de la platine, des composants optiques, des obturateurs, des filtres et des détecteurs, déjà couramment utilisé, permet de telles manipulations lors d'expériences qui étaient tout simplement impossibles pour les humains travaillant sur des microscopes mécaniques. L'utilisation croissante de l'optoélectronique en microscopie à fluorescence a conduit au développement de pinces optiques pour la manipulation de structures et de particules subcellulaires, l'observation de molécules individuelles et une large gamme d'applications spectroscopiques sophistiquées.
Combinaisons de filtres fluorescents
Des combinaisons de filtres d'interférence et d'absorption d'épifluorescence sont placées dans des cubes filtrants (ou unités optiques) et comprennent un filtre d'excitation, un séparateur de faisceau dichroïque (souvent appelé miroir) et un filtre de blocage (ou d'émission), comme le montre la figure 1. (un). Ce guide peut être utile pour sélectionner des combinaisons de filtres qui correspondent aux caractéristiques spectrales d’absorption et d’émission des chromophores utilisés en microscopie à fluorescence à grand champ. Les courbes spectrales d'une combinaison typique de filtres passe-bande hautes performances dans la plage d'excitation bleue sont illustrées à la figure 1 (b). Les combinaisons de filtres de fluorescence Nikon sont disponibles avec des filtres d'excitation à bande étroite, moyenne et large bande et leurs filtres d'émission spécifiques ou à large bande correspondants.

Riz. 8. Courbes spectrales d'un bloc de filtres de lumière fluorescente
Excitation ultraviolette - L'ensemble de filtres de fluorescence d'excitation UV de Nikon comprend quatre combinaisons soigneusement équilibrées qui incluent des filtres passe-bande conventionnels ou des filtres d'émission à large bande (blocage) qui peuvent transmettre sélectivement l'émission fluorescente dans une plage étroite ou large des parties bleues, vertes et rouges du spectre visible. Ces combinaisons de filtres couvrent la plage d'excitation de 330 à 380 nanomètres avec des bandes passantes de 10, 40 et 50 nanomètres. Trois combinaisons utilisent le même miroir dichroïque et la quatrième a une longueur d'onde de coupure inférieure pour correspondre à la bande d'excitation étroite. Les combinaisons de filtres UV comprennent des filtres d'émission à bande passante fixe ou à large bande.
Excitation violette : le jeu de filtres fluorescents à excitation violette de Nikon comprend trois combinaisons comprenant des filtres passe-bande ou d'émission à large bande (blocage) conventionnels qui peuvent transmettre sélectivement l'émission fluorescente dans une plage étroite ou large de régions bleues, vertes et rouges du spectre. Ces combinaisons de filtres couvrent la plage d'excitation de 379 à 420 nanomètres avec des bandes passantes de 10, 22 et 40 nanomètres. Deux combinaisons utilisent le même miroir dichroïque et la troisième a une longueur d'onde de coupure inférieure pour correspondre à la bande d'excitation aux longueurs d'onde plus courtes.
Excitation bleu-violet - L'ensemble de filtres de fluorescence d'excitation bleu-violet de Nikon comprend quatre combinaisons comprenant des filtres passe-bande conventionnels ou des filtres d'émission à large bande (blocage) qui peuvent transmettre sélectivement l'émission fluorescente dans une plage étroite ou large des régions bleues, vertes et rouges du visible. spectre. Ces jeux de filtres supplémentaires couvrent la plage d'excitation de 400 à 446 nanomètres avec des bandes passantes de 10, 20 et 40 nanomètres. Trois combinaisons utilisent le même miroir dichroïque, tandis que la quatrième a une longueur d'onde de coupure plus élevée (de 5 nanomètres) pour correspondre aux autres composants.
Excitation bleue : le jeu de filtres fluorescents à excitation bleue de Nikon se compose de six combinaisons équilibrées comprenant des filtres passe-bande ou d'émission à large bande (blocage) conventionnels qui peuvent transmettre sélectivement l'émission fluorescente dans une plage étroite ou large des régions vertes, jaunes, rouges et infrarouges du spectre. . Ces combinaisons de filtres couvrent la plage d'excitation de 420 à 495 nanomètres avec des bandes passantes de 20, 30, 40 et 70 nanomètres. Cinq combinaisons utilisent le même miroir dichroïque et la sixième a une longueur d'onde de coupure inférieure pour augmenter le signal reçu. Tous les filtres coupe-bande pour les kits de filtration par excitation bleue Nikon ont une largeur spectrale de 40 nanomètres. L'un des filtres (B-3A) est conçu pour être utilisé avec un éclairage à lampe halogène au tungstène.
Excitation verte - Le jeu de filtres fluorescents à excitation verte de Nikon se compose de six blocs, qui comprennent des filtres passe-bande ou d'émission à large bande (blocage) conventionnels capables de transmettre sélectivement l'émission fluorescente dans une plage étroite ou large de régions jaunes, oranges, rouges et proches infrarouges du spectre. Ces combinaisons de filtres couvrent une plage d'excitation de 510 à 560 nanomètres avec des bandes passantes de 10, 25, 30 et 50 nanomètres (y compris des bandes d'excitation étroites, moyennes et larges). Trois combinaisons utilisent le même miroir dichroïque (565 nanomètres) et les trois autres ont une longueur d'onde de coupure plus longue (570 et 575 nanomètres). Deux des six kits de filtres d'excitation verts de Nikon comprennent des filtres coupe-bande de 60 et 75 nanomètres.
Excitation jaune - Le jeu de filtres fluorescents à excitation jaune de Nikon se compose de deux combinaisons équilibrées comprenant des filtres d'émission (blocage) avec une bande passante spécifique, capables de transmettre sélectivement l'émission fluorescente dans les régions orange et rouge du spectre. Ces combinaisons de filtres optionnelles couvrent la plage d'excitation de 532 à 587 nanomètres avec des bandes passantes de 40 et 55 nanomètres. Les deux combinaisons incluent le même miroir dichroïque (avec une coupure à 595 nanomètres). Les deux kits de filtration d'excitation jaune de Nikon comprennent des filtres coupe-bande de 60 et 75 nanomètres.
Excitation rouge - La combinaison de filtres fluorescents à excitation rouge de Nikon est représentée par une seule unité, qui comprend un filtre d'émission passe-bande (blocage) capable de transmettre sélectivement l'émission fluorescente dans les régions du rouge lointain et du proche infrarouge du spectre. Le centre de la bande passante du filtre bloquant est de 700 nanomètres et sa largeur est de 75 nanomètres (de 663 à 738 nanomètres). La large bande d'excitation de 60 nm, de 590 à 650 nm, couvre les longueurs d'onde orange et rouge. La combinaison Cy5 HYQ comprend un miroir dichroïque avec une coupure à 660 nanomètres, soit 10 nanomètres plus élevée que la coupure de la bande d'excitation.
Excitation de la protéine fluorescente jaune (YFP) - Pour YFP, Nikon a développé une combinaison équilibrée de haute qualité qui améliore les capacités de détection des protéines fluorescentes (fournies par trois kits de filtres de protéine fluorescente verte (GFP)) en utilisant des filtres pour les variantes de GFP à longueur d'onde plus longue ( YFP et EYFP). La banque de filtres YFP HYQ comprend des filtres d'excitation et d'émission (arrêt) avec des bandes passantes relativement étroites conçues spécifiquement pour correspondre aux caractéristiques spectrales de la protéine fluorescente jaune améliorée par YFP (YFP), permettant d'évaluer la fluorescence des dérivés de YFP séparément des autres protéines fluorescentes.
Excitation double bande - L'ensemble de filtres de fluorescence double bande de Nikon se compose de trois combinaisons soigneusement équilibrées de filtres double bande (filtres d'excitation et d'émission (coupure)) combinés en une seule unité qui transmet simultanément de manière sélective l'émission fluorescente de deux fluorophores. Chacune des unités de filtration est combinée de manière optimale avec une paire de fluorochromes spécifique, bien qu'elle puisse également fonctionner efficacement avec d'autres paires de colorants fluorescents ayant les mêmes profils spectraux d'absorption et d'émission. Grâce à une adaptation précise des bandes, avec des transitions abruptes de bande à bande entre les régions de réflexion et de transmission, les différents signaux d'excitation et d'émission sont séparés avec un minimum d'interférences superposées.
Excitation à trois bandes - Les filtres fluorescents à trois bandes Nikon sont représentés par deux blocs équilibrés, comprenant des filtres à trois bandes (filtres d'excitation et d'émission (blocage)), transmettant sélectivement l'émission fluorescente de trois fluorophores simultanément. Chacune des unités de filtration est combinée de manière optimale avec un ensemble spécifique de trois fluorochromes, bien qu'elle puisse également fonctionner efficacement avec d'autres combinaisons de colorants présentant les mêmes profils spectraux d'absorption et d'émission. Grâce à une adaptation précise des bandes, avec des transitions interbandes abruptes entre les régions de réflexion et de transmission, les différents signaux d'excitation et d'émission sont séparés avec une interférence minimale entre eux. Triples cubes.
Cubes HYQ - Les combinaisons de filtres de fluorescence HYQ de Nikon comportent quatre blocs de haute qualité soigneusement équilibrés, chacun intégrant des filtres d'émission passe-bande (blocage) pour transmettre sélectivement la fluorescence dans une plage limitée. Chaque désignation de filtre HYQ reflète le nom du fluorochrome pour lequel il a été conçu, mais dans ses plages d'excitation, chaque combinaison peut être utilisée pour observer différents fluorochromes avec des caractéristiques correspondantes.
Liste de base des unités de filtrage fluorescentes Nikon
Les conventions de dénomination de Nikon sont basées sur des termes mixtes apparus au début des années 1990. À cette époque, toutes les combinaisons de filtres supplémentaires de Nikon étaient produites à l'aide de la méthode de revêtement dur, mais aujourd'hui, de nombreux filtres sont fabriqués à l'aide de méthodes avancées de revêtement doux. Bien que les revêtements souples soient plus sensibles à l’humidité et à la chaleur et nécessitent une manipulation plus soigneuse (par rapport aux revêtements durs), ils présentent des densités optiques plus élevées et facilitent davantage le réglage précis de bandes passantes spécifiques. Comprendre les conventions des combinaisons de filtres Nikon vous permet de sélectionner rapidement les filtres nécessaires pour des fluorophores spécifiques.
La première lettre du système de notation alphanumérique de Nikon indique la région du spectre d'excitation (par exemple, UV, V, B et G sont des abréviations des mots anglais "ultraviolet" - ultraviolet, "violet" - violet, "blue" - bleu , et « vert » - vert, respectivement). Le chiffre suivant le codage du spectre d'excitation indique la bande passante du filtre d'excitation : 1 correspond à une excitation bande étroite, 2 à une excitation bande moyenne et 3 à une excitation large bande. Enfin, une ou plusieurs lettres suivant le chiffre correspondant à la bande passante d'excitation indiquent les caractéristiques du filtre bloquant. La lettre A indique un filtre de coupure à large bande standard avec la longueur d'onde de coupure la plus basse, B indique un filtre d'émission à large bande ayant une longueur d'onde de coupure plus élevée. La désignation E (de l'anglais « Enhanced » - Enhanced) dans les filtres d'émission passe-bande indique des performances améliorées dans le sens de réduction de l'interaction interférentielle des signaux séparés. La désignation E/C indique une combinaison de revêtements interférentiels doux conçus spécifiquement pour fonctionner avec des colorants spécifiques tels que le DAPI, le FITC, le TRITC et le Texas Red.
MICROSCOPIE LUMINESCENTE(du latin lumen, luminis _ light ; grec, mikros small + skopeo considérer, explorer ; syn. microscopie à fluorescence) - une méthode de microscopie qui permet d'observer la luminescence primaire ou secondaire des micro-organismes, cellules, tissus ou structures individuelles qui les composent. La luminescence (voir) est excitée par la partie à ondes courtes (bleu-violet) de la lumière visible ou des rayons ultraviolets dont la longueur d'onde est proche de la lumière visible. La couleur de la luminescence, c'est-à-dire la longueur d'onde de la lumière émise, dépend de structure chimique et sur l'état physico-chimique de l'objet microscopique, qui permet d'utiliser L. m à des fins de diagnostic microbiologique et cytologique, pour différencier les composants individuels de la cellule. La luminescence primaire est inhérente à un certain nombre de facteurs biologiques substances actives, tels que les acides aminés aromatiques, les porphyrines, la chlorophylle, les vitamines A, B2, B1, certains antibiotiques (tétracycline) et les substances chimiothérapeutiques (akrikhin, rivanol). La luminescence secondaire ou induite résulte du traitement d'objets microscopiques avec des colorants fluorescents - les fluorochromes. Certains de ces colorants sont distribués de manière diffuse dans les cellules (par exemple la fluorescéine), d'autres se lient sélectivement à certaines structures cellulaires ou même à certains produits chimiques. substances. Cette capacité de coloration sélective des fluorochromes permet une cytologie fluorescente. et cytochimie fluorescente. recherche.
Dans l'histoire du développement de L. m. il y a plusieurs étapes associées à l'amélioration de la technique :
1) la preuve par A. Köhler de la possibilité fondamentale de créer un microscope à luminescence ; 2) la création en 1911 d'un microscope à luminescence, utilisé par le botaniste russe M. S. Tsvet pour étudier la luminescence de la chlorophylle cellules végétales; 3) l'utilisation de solutions très diluées de fluorochromes se liant sélectivement à certaines structures cellulaires [Haitinger (M. Haitinger, 1933-1935)], et surtout d'acridine orange [Haitinger, S. Strugger, 1940] ; 4) développement d'une méthode d'excitation de la luminescence par la lumière incidente à travers une lentille de microscope utilisant une plaque de séparation de faisceau interférentiel (E. M. Brumberg et G. N. Krylova, 1953) et production de microscopes et de dispositifs à luminescence basés sur ce principe par l'industrie nationale (ML -1, ML-2, OI-17); 5) création de la méthode d'immunofluorescence (voir), qui a trouvé de larges applications en microbiologie, en immunologie et dans d'autres domaines de la biologie médicale. recherche [Koons (A. N. Goons), 1942, 1950].
En URSS, contribution significative au développement et à la diffusion de la médecine médico-biologique. recherche effectuée par M. N. Meisel.
Pour réaliser la LM, on utilise soit des microscopes luminescents spéciaux, soit des accessoires pour microscopes biologiques conventionnels, leur permettant d'être utilisés pour observer la luminescence de microobjets (voir Microscope). La conception des microscopes à fluorescence repose sur des principes physiques lois de la luminescence. L'une d'elles est la loi de Stokes, selon laquelle le maximum du spectre de luminescence est décalé vers la région des ondes longues par rapport au spectre de la lumière excitatrice. Cela permet d'utiliser le principe des filtres de lumière croisée pour la microscopie laser, qui consiste dans le fait que le rayonnement lumineux à ondes courtes (ultraviolet, bleu-violet), qui excite la luminescence, est émis par un filtre de lumière excitatrice placé devant l'illuminateur du microscope. Après avoir traversé le médicament, la luminescence est excitée, cette lumière est complètement absorbée par un filtre bloquant qui transmet la lumière de luminescence à longueur d'onde plus longue.
Un microscope à luminescence est équipé d'une puissante source d'éclairage à haute luminosité de surface, dont l'émission maximale se situe dans la région des ondes courtes du spectre visible, d'un système de filtres de lumière, ainsi que d'une plaque séparatrice de faisceau interférentiel (ou d'un ensemble de telles plaques) utilisé pour exciter la luminescence par la lumière incidente. Ce système d'excitation de luminescence par lumière incidente à travers un illuminateur opaque, utilisé dans les microscopes à luminescence domestiques (et dans dernièrement et dans les microscopes à luminescence produits par des sociétés étrangères), présente un certain nombre d'avantages significatifs : 1) une plaque de séparation de faisceau d'interférence sur laquelle sont déposées des couches de diélectriques réfléchit sélectivement plus de 90 % de la lumière qui excite la luminescence sur l'échantillon, et presque complètement transmet une lumière de luminescence de longueur d'onde plus longue, ce qui permet d'augmenter la luminosité de luminescence ; 2) la lentille du microscope sert simultanément de condenseur pour le système d'éclairage ; par conséquent, lors de l'utilisation d'objectifs à immersion à grande ouverture et à fort grossissement, l'éclairage de la préparation et, par conséquent, la luminosité de la luminescence augmentent proportionnellement à la quatrième puissance de l'ouverture de l'objectif ; 3) La microscopie à fluorescence peut être combinée avec la microscopie à contraste de phase et à interférence lorsqu'elle est éclairée par le bas à travers le condenseur du microscope. Les sources d'éclairage pour LM sont souvent des lampes à mercure-quartz à ultra haute pression, ainsi que des lampes au xénon et des lampes à incandescence quartz-halogène. Du verre optique coloré dans la masse est utilisé comme filtre de lumière, ou des filtres interférentiels présentant de meilleures caractéristiques spectrales sont utilisés.
Pour exciter la luminescence pendant le LM, des générateurs quantiques optiques – des lasers – sont également utilisés, dont le rayonnement est très intense et monochromatique. Dans ce cas, il n’est pas nécessaire d’utiliser des filtres de lumière excitants.
Étant donné que les régions ultraviolettes de grande longueur d'onde, bleu-violet et parfois vertes du spectre sont généralement utilisées pour exciter la luminescence dans les microscopes à luminescence, les optiques en verre ordinaires et les lames et lamelles de verre ordinaires, qui transmettent le rayonnement dans cette partie du spectre et n'ont pas leur propre luminescence, sont utilisés dans un microscope à luminescence. Les milieux d’immersion et de confinement doivent également répondre à ces exigences.
Le tampon peut être utilisé comme milieu de confinement pour les médicaments. solution de glycérine, ainsi que des polymères non luminescents (polystyrène, alcool polyvinylique, etc.).
Parallèlement à l'évaluation visuelle de l'image microscopique luminescente, la microphotographie est utilisée (voir Microphotographie). La microphotographie luminescente présente un certain nombre de fonctionnalités. D'une part, une luminosité insuffisante de la lueur nécessite une longue exposition, d'autre part, sous l'influence d'une lumière excitante, l'intensité de la luminescence diminue rapidement, les préparations s'estompent, les cellules vivantes sont endommagées et meurent, et il est donc impossible d'enregistrer la dynamique des processus se produisant dans les cellules. Pour surmonter ces difficultés, il est nécessaire d'utiliser du matériel photographique à haute sensibilité spectrale générale et sélective, des objectifs à grande ouverture, des oculaires à grossissement intrinsèque minimal, mais suffisant pour transmettre les détails de l'objet, et des accessoires microphotographiques de petit format. Il est également possible de traiter les préparations avec des substances réduisant la décoloration (hydroquinone, etc.).
M. Ya Korn et M. M. Butlov et al. (1968) ont développé des équipements et des techniques de microphotographie et de tournage luminescents couleur utilisant des amplificateurs de luminosité électro-optiques, qui permettent de réduire l'exposition de plusieurs ordres de grandeur et d'enregistrer la dynamique des processus se produisant dans les cellules vivantes.
Pour enregistrer quantitativement l'intensité de luminescence des structures de microobjets - cytofluorimétrie - des méthodes principalement photoélectriques sont utilisées en utilisant un tube photomultiplicateur (PMT) comme dispositif d'enregistrement sensible (voir Photomultiplicateurs). Ils utilisent également une méthode pour enregistrer l'intensité de luminescence des cellules et de leurs structures dans certaines parties du spectre : la cytospectrofluorimétrie. Les avantages de la cytofluorimétrie par rapport à la cytophotométrie d'absorption sont sa sensibilité plus élevée et l'absence d'influence de la nature de la distribution de la substance dans la cellule ou de la structure cellulaire sur les résultats de mesure.
L'une des options pour l'enregistrement quantitatif de la luminescence des microobjets est la méthode de cytofluorimétrie pulsée (cytofluorimétrie en flux) et le « tri » des cellules selon les caractéristiques luminescentes. Ces méthodes permettent d'analyser l'intensité de luminescence de dizaines de milliers de cellules par minute et de les séparer selon la nature de la luminescence. Parmi les méthodes d'étude luminescente des microobjets, les plus utilisées sont la fluorochromisation directe - coloration au fluorochrome et immunofluorescence.
L'industrie nationale produit divers équipements pour la microscopie microscopique. Le microscope le plus utilisé est le ML-2, produit en plusieurs versions. Une série de microscopes fluorescents unifiés «Lumam» est également produite, ainsi que des éclairages fluorescents avec lampes halogènes à quartz (OI-28, OI-30).
Les microscopes de la série "Lumam" sont constitués d'unités unifiées dont diverses combinaisons permettent d'obtenir trois modèles de travail (P1 - RZ) et trois modèles de recherche (I1 - I3), différant les uns des autres par leur configuration et leurs possibilités d'utilisation ( Figue.).
Les illuminateurs OI-28 et OI-30 sont installés sur les microscopes biologiques conventionnels ; ils sont conçus pour éclairer les objets d'en haut à travers un hublot opaque avec une lumière qui suscite une luminescence visible.
L'illuminateur OI-30 se distingue par le fait que son kit comprend des lentilles de contact.
L. m. sont largement utilisés en virologie, microbiologie, hématologie, coin, cytodiagnostic (notamment en oncologie pour la détection des cellules malignes), en cytogénétique pour l'étude des chromosomes. A cet effet, le fluorochrome orange d'acridine est utilisé depuis longtemps ; Les fluorochromes bromure d'éthidium (bromure d'éthidium) et iodure de propidium (iodure de pripidium) sont également utilisés, principalement pour la cytofluorimétrie de l'ADN, ainsi que des versions luminescentes de la réaction de Feilgen. L'acridine orange s'est répandue en microscopie à luminescence des nucléoprotéines en raison du fait que les complexes formés par ce fluorochrome avec de l'ADN double brin ont une luminescence verte et que les complexes avec de l'ARN et de l'ADN simple brin ont une luminescence rouge. Des méthodes cytochimiques luminescentes sont également connues. méthodes d'identification des protéines et des lipides. Pour l'étude qualitative et quantitative de la localisation des protéines dans les cellules, des colorants procion, de la fluorescamine sont utilisés et des lipides - 3,4-benzpyrène, phosphine ZR, etc. La tétracycline et ses dérivés sont utilisés pour l'étude microscopique luminescente du tissu osseux et de certains changements dans les cellules lors d'une tumeur maligne.
L. m. en combinaison avec la fluorochromisation directe de préparations fixes est utilisée dans le diagnostic bactérioscopique pour la détection des mycobactéries acido-résistantes, des gonocoques, des agents pathogènes de la diphtérie, des agents pathogènes du paludisme dans les frottis sanguins, etc. Les avantages de cette méthode résident dans sa sensibilité plus élevée que celle aux méthodes de coloration conventionnelles (par exemple, coloration de Ziehl-Neelsen). Le placage au fluorochrome est également utilisé dans les bactéries sanitaires. recherche pour la détection et le dénombrement de micro-organismes dans l'eau et le sol. L'une des méthodes de diagnostic microscopique à fluorescence est la détection de microcolonies sur des filtres à membrane après une croissance à court terme et une fluorochromisation.
En 1940, Strugger a proposé d'utiliser l'acridine orange pour différencier les bactéries vivantes et mortes, mais des études ultérieures ont montré fiabilité insuffisante cette méthode. À cet égard, pour déterminer la viabilité des cellules (en particulier lorsqu'elles sont exposées à des facteurs cytotoxiques), le diacétate de fluorescéine ou sa combinaison avec le bromure d'éthidium est utilisé. L'ester non luminescent de fluorescéine est clivé par les estérases d'une cellule viable, libérant de la fluorescéine verte brillamment luminescente, et le bromure d'éthidium provoque une luminescence rouge uniquement dans les cellules mortes.
Etudier la physique et la chimie l'état des membranes cellulaires est ce qu'on appelle. échantillons fluorescents hydrophobes. À cette fin, les cellules sont traitées avec des substances qui ne luminescentes pas en solution, mais commencent à luminescence en se liant aux zones hydrophobes des membranes cellulaires, et l'intensité et la couleur de la luminescence dépendent du produit chimique. structure et physico-chimique états des structures auxquelles ces fluorochromes sont associés. L’une des substances les plus courantes de ce type est l’acide 1-anilino-S-naphtalène sulfonique (1,8 ANS).
Utiliser la méthode d'étude physique et chimique Les états des macromolécules, auxquels les fluorochromes sont associés dans diverses structures cellulaires, sont également obtenus par microscopie à fluorescence polarisée.

Un avantage significatif de la LM par rapport aux autres méthodes d’examen microscopique est la possibilité de fluorochromisation intravitale (couleur Fig. 1-3) et supravitale utilisant des concentrations très faibles et peu toxiques de fluorochromes. Dans ce cas, différents fluorochromes peuvent se lier à différentes structures cellulaires. L'acridine orange, par exemple, s'accumule dans les lysosomes d'une cellule vivante et ils commencent à émettre une fluorescence avec la lumière rouge. Les bactéries phagocytées à l’intérieur des phagosomes acquièrent la même luminescence. La tétracycline se lie aux mitochondries des cellules ou à leurs analogues dans les bactéries et luminescente avec une lumière jaune-vert, et l'intensité de la luminescence (la quantité de tétracycline liée) dépend de la sensibilité des bactéries à cet antibiotique.
Il est également possible de fluorochromiser des cellules et des tissus in situ pour les étudier à l'aide de contact L. m.
La microscopie à fluorescence des virus est utilisée en laboratoire. diagnostic de maladies virales pour identifier l'antigène viral dans les cellules, étude de la chimie. composition des inclusions virales intracellulaires, détermination de la concentration relative des antigènes viraux et acide nucléique par l'intensité de la fluorescence spécifique, etc. Selon les objectifs de l'étude, des frottis, des empreintes, des grattages de tissus ou des préparations de culture cellulaire sont utilisés comme objets.
En virologie moderne, les deux méthodes de LM les plus courantes sont : 1) l'identification et la différenciation des acides nucléiques ensemble de virus dans les cellules infectées et les suspensions virales purifiées à l'aide de fluorochromes d'aminoacridine ; 2) détection d'antigènes viraux par immunofluorescence.
Parmi les aminoacridines, l'acridine orange est le plus souvent utilisée, ce qui donne aux molécules d'acides nucléiques double brin (généralement de l'ADN) une fluorescence verte et d'acides nucléiques simple brin (généralement de l'ARN) rouge rubis (couleur Fig. 4-8). La méthode est applicable aussi bien sur des préparations natives qu'après fixation dans l'acétone, le liquide de Carnoy, etc. Les résultats de la coloration dépendent largement de la concentration (généralement 1 : 10 000 - 1 : 100 000) et du pH du fluorochrome.
La méthode d'immunofluorescence est utilisée pour identifier les virus dans les cellules, étudier la dynamique d'accumulation d'antigène viral dans une cellule, déterminer la localisation intracellulaire des accumulations de virus, déterminer la nature des inclusions virales, étudier la structure antigénique des virus, différencier des virus étroitement apparentés, titrer les virus dans les cultures cellulaires, identifier les antigènes des virus tumoraux dans les tissus et les cellules, étudier la pathogenèse des maladies virales, surveiller la contamination virale des cultures cellulaires, étudier le hron, les infections virales, etc. La méthode est applicable dans les modifications directes et indirectes. Pour interpréter correctement les résultats de l'étude, il est important de prendre en compte la concentration et la pureté des anticorps utilisés et de leurs conjugués avec les fluorochromes, le moment et la température de fixation des médicaments (généralement avec de l'acétone) et leur traitement avec des anticorps. Le plus souvent, l'isothiocyanate de fluorescéine (FITC), qui donne une lueur verte caractéristique (couleur Fig. 9), et le sulfochlorure de lissamine-rhodamine B 200, qui brille en rouge orangé, sont utilisés pour marquer les anticorps.
La méthode d’immunofluorescence permet également la détection d’anticorps dirigés contre des antigènes viraux dans les cellules et les tissus.
L. m. des virus est utilisé pour diagnostiquer des infections telles que la variole, l'herpès, les oreillons, etc. L. m. revêt une importance particulière dans le diagnostic express des infections virales respiratoires, lorsque des empreintes de la muqueuse nasale des patients (rhinocytogrammes) sont traité en utilisant les méthodes ci-dessus suivantes dans le but d'identifier les antigènes ou de déterminer le type d'acide nucléique. Que. effectuer un diagnostic différentiel entre les infections causées par les virus grippaux A2 ou B, le parainfluenza, les adénovirus, le virus respiratoire syncytial ou des combinaisons de ces virus. Le diagnostic par LM de cultures cellulaires infectées par du matériel provenant de patients est également possible. Le diagnostic final dans tous les cas est posé en combinant les données de L. m. avec les résultats des études virales et séroliennes des patients.
La microscopie luminescente des organes et des tissus est l'une des méthodes modernes recherche utilisée en histologie normale et pathologique. Les principaux avantages de L. m sont une sensibilité élevée (au moins 1 000 fois plus sensible que les méthodes cytochimiques et histochimiques conventionnelles), la facilité mesure quantitative contenu de divers produits chimiques. composants tissulaires et cellules, disponibilité des équipements. Pour la luminescence des organes et des tissus, des luminescences primaires et secondaires sont utilisées. La luminescence primaire (lueur luminescente qui se produit sans prétraitement médicamenteux) est possédée avec une intensité suffisante par certaines substances qui composent les cellules et les tissus : vitamines (la vitamine B2 donne une luminescence jaune-vert, la vitamine B1 dans une solution alcaline se transforme en trichrome et donne luminescence bleue, le carotène luminescent avec une lumière jaune-vert, la vitamine A, lorsqu'elle est irradiée dans le spectre UV, a une luminescence bleu-blanc), les hormones (les œstrogènes, l'adrénaline donnent une luminescence jaune-vert, la sérotonine, la noradrénaline, lorsque les médicaments sont traités avec des vapeurs de acide sulfurique concentré, ils ont une luminescence jaune) , lipopigments (la lipofuscine donne une luminescence rouge, céroïde - bleuâtre), etc. Le principe de la luminescence primaire est à la base de la cytochimie, une étude quantitative du contenu de divers composants cellulaires (principalement des protéines) à l'aide la méthode de luminescence dans les rayons UV.
La luminescence secondaire des organes et des tissus est obtenue en traitant des préparations avec des fluorochromes (voir). L'acridine orange est utilisée pour diagnostiquer le cancer dans les préparations de tsitol et de gistol. Le même colorant est utilisé pour déterminer les premiers stades de l’infarctus du myocarde (les zones d’ischémie ont une luminescence vert-jaune). La corifosphine et l'acridine orange sont utilisées pour identifier les mucopolysaccharides acides. Les fluorochromes tels que la caféine 5 et la rhodamine peuvent être utilisés pour déterminer le glycogène. La phosphine 3P est utilisée pour déterminer les lipides ; dans le même but, une solution de 3,4-benzpyrène dans une solution saturée de caféine est utilisée (les lipides ont une luminescence blanc bleuâtre). La thioflavine T.S. colore l'amyloïde (luminescence verte), elle est donc largement utilisée pour le diagnostic de l'amylose. organes internes. À l'aide d'une solution de morin dans l'alcool, le calcium dans les tissus est déterminé (luminescence verte). Lors du traitement de préparations avec une solution de noir solochrome, il est possible de détecter de l'aluminium (luminescence jaune-orange). Grâce à la rhodamine 6G, le tensioactif est dosé dans les poumons (luminescence orange).
Grâce à la méthode d'immunofluorescence, les hormones, les antigènes et les anticorps peuvent être détectés (couleur Fig. 9), divers produitséchange, identification de tumeurs histogénétiquement immatures, diverses informations. maladies, etc Le développement de l’immunochimie a encore élargi les capacités de cette méthode. Il est devenu possible de déterminer des substances non protéiques dans les tissus et les cellules à l'aide d'haptènes artificiels.
Bibliographie: Barsky I. Ya., Polyakov N. I. et Yakubenas V. A. V. Microscopie de contact, M., 1976, bibliogr.; Ershov F.I. Détection microscopique luminescente des changements précoces dans les acides nucléiques et les lipides dans les cellules infectées, Vopr, virusol., .No 1, p. 3, 1964, bibliogr.; 3élénine A.V. Interaction des dérivés aminés de l'acridine avec la cellule, M., 1971, bibliogr. ; 3 chez b-zhitsky Yu. Méthode de microscopie à luminescence, L., 1964, bibliogr.; K a r-mysheva V.Ya. Application de la méthode des anticorps fluorescents en virologie, M., 1979 ; M e y s e l M. N. Microscopie fluorescente et cytochimie en microbiologie générale, dans l'ouvrage : Usp. microbiol., éd. A. A. Imshenetsky, vol 7, p. 3, M., 1971 ; Mikhailov I. F. et Dyakov S. I. Microscopie luminescente, M., 1961, bibliogr.; Strukov A. I. et Kondratyev V. S. Méthode microscopique luminescente en pratique pathologique, Arkh. pathol., t. 28, n° 8, p. 77, 1966, bibliogr.; Fridman I. A. et Kustarov N. P. Études cytologiques luminescentes dans la pratique obstétricale et gynécologique, L., 1974, bibliogr.; Automatisation en microbiologie et immunologie, éd. par C.-G. Il avait un. T. Illeni, N.Y., 1975 ; L'automatisation de la cytologie du cancer de l'utérus, éd. par G. L. Wied a.o., Chicago, 1976 ; Avec une personne T. a.o. Fluorochromes liant l'ADN pour l'étude de l'organisation du noyau métaphasique, Exp. Cell Res., v. 58, p. 141, 1969 ; Techniques de fluorescence en biologie cellulaire, éd. par A.A. Thaer a. M. Sernetz, New York, 1973 ; Vaillier J. a. Vaillier D. Caractérisation des sous-populations cellulaires du thymus par une sonde fluorescente hydrophobe, le l-anilino-8-naph-thalène sulfonate, Clin. exp. Immunol v. 30, p. 283, 1977.
M. Ya. V. A. Varshavsky (pat. an.), Ya. E. Khesin (vir.).
) — une méthode de détection de microobjets fluorescents à l'aide d'un microscope optique (optique). Largement utilisé dans les domaines de la science des matériaux et du biomédical.
Description
Matériel biologique, en règle générale, émet une fluorescence extrêmement faible en soi, mais grâce à l'utilisation de molécules fluorescentes brillantes et diverses (fluorophores) capables de colorer spécifiquement différentes structures et structures tissulaires, la méthode de microscopie à fluorescence s'est avérée très précieuse pour les sciences biomédicales .
Les méthodes traditionnelles de microscopie à fluorescence ont une résolution nettement inférieure à celle ou. Cependant, contrairement à cette dernière, la microscopie optique permet d'observer la microstructure interne des cellules et même des petits organismes, non seulement fixes, mais aussi vivants. Grâce à cela, la microscopie à fluorescence s'est avérée être la meilleure méthode pour étudier les mécanismes de fonctionnement des organismes au niveau cellulaire, subcellulaire et niveaux moléculaires.
Un microscope à fluorescence se compose d'une source de lumière qui excite un fluorophore ; un détecteur qui enregistre le rayonnement fluorophore ; système optique qui concentre la lumière et agrandit un objet. D'après les travaux classiques d'E. Abbe, la résolution d'un système optique basé sur l'utilisation de lentilles est limitée par la propriété de diffraction de la lumière. La distance maximale à laquelle deux objets peuvent être distingués ( d), déterminé par la longueur d'onde de la lumière, l'ouverture angulaire de la lentille et l'indice de réfraction du milieu n:
Parce que d'habitude n < 1,56, < 70 o , а длина волны используемого излучения находится в диапазоне 350–600 нм, то лучшее разрешение традиционных составляет более 200 нм в фокусной плоскости и более 450 нм вдоль оптической оси.
Le développement intensif de la microscopie à fluorescence au tournant des 20e et 21e siècles a conduit au développement de nouvelles méthodes - ainsi qu'à un certain nombre d'approches qui ont permis de surmonter la barrière de diffraction de la résolution optique et d'obtenir une nano-résolution sans précédent. ().
Auteur
- Borisenko Grigori Gennadievitch
Sources
- Kassens M. et al. Bases de la microscopie optique et de l'imagerie. -GIT Verlag GmbH & Co. KG, 2006. - 52 p.
- Abbe, E. Beiträge zur Theorie des Mikroskops und der Mikroskopischen Wahrnehmung // Arch. Microsc. Anat. Entwicklungsmech. 1873. Bd. 1873. 9. Articles 413 à 468.
Riz. 1. Microscope à épifluorescence
La fluorescence a été décrite pour la première fois en 1852 par le scientifique britannique George Stokes, qui a inventé le terme alors qu'il menait des expériences avec la fluorite (spath fluor), qui émet une lumière rouge lorsqu'elle est irradiée par une lumière ultraviolette. Stokes a remarqué que la longueur d'onde de l'émission fluorescente est toujours supérieure à la longueur d'onde de la lumière d'excitation. Les premières recherches menées au XIXe siècle ont montré que de nombreux échantillons (notamment des minéraux, des cristaux, des résines, des matières premières médicinales, des huiles, de la chlorophylle, des vitamines et des composés inorganiques) émettent une fluorescence lorsqu'ils sont irradiés par une lumière ultraviolette. Cependant, l’utilisation des fluorochromes dans la recherche biologique pour colorer les composants tissulaires, les bactéries et autres agents pathogènes n’a commencé que dans les années 1930. Certains de ces colorants étaient extrêmement spécifiques et ont stimulé le développement de la microscopie à fluorescence.En raison de plusieurs fonctionnalités difficiles à réaliser avec la microscopie optique traditionnelle à contraste amélioré, la microscopie à fluorescence est devenue un outil important dans la recherche biologique et biomédicale et dans la science des matériaux. L'utilisation d'ensembles de fluorochromes a permis d'isoler des cellules très spécifiques et des composants cellulaires submicroscopiques de substances non fluorescentes. Avec un microscope à fluorescence, même des molécules individuelles peuvent être détectées. Grâce à la multicoloration fluorescente, différents colorants peuvent identifier plusieurs molécules cibles simultanément. Bien que la résolution spatiale d'un microscope à fluorescence soit limitée en dessous par la limite de diffraction, qui dépend des caractéristiques spécifiques de l'échantillon, la détection de molécules fluorescentes en dessous de cette limite est tout à fait possible.
De nombreux échantillons présentent une autofluorescence lorsqu’ils sont irradiés (sans utilisation de fluorochromes), et ce phénomène est largement utilisé en botanique, en pétrologie et dans l’industrie des semi-conducteurs. En revanche, l’étude des tissus d’animaux ou d’agents pathogènes est souvent compliquée par une autofluorescence non spécifique extrêmement faible ou, à l’inverse, forte. Bien plus importante dans ce cas est l'introduction dans les tissus de fluorochromes (ou fluorophores), excités à une certaine longueur d'onde et émettant de la lumière avec l'intensité requise. Les fluorochromes sont des colorants qui, se fixant indépendamment sur des structures visibles ou invisibles, ont une sélectivité élevée envers les cibles et un rendement quantique élevé (le rapport entre le nombre de photons émis et le nombre de photons absorbés). La croissance rapide de l’utilisation de la microscopie à fluorescence est étroitement liée à l’émergence de nouveaux fluorophores synthétiques et naturels qui présentent des profils d’intensité d’excitation et d’émission spécifiques et sont « ciblés » sur des cibles biologiques données.
Bases des processus d'excitation et d'émission
Le principe de fonctionnement d'un microscope à fluorescence consiste à irradier un échantillon à des longueurs d'onde dans la plage requise et définie avec précision, suivi de la séparation de la fluorescence émise beaucoup plus faible du flux de lumière excitatrice. Dans un microscope bien réglé, seule la lumière émise doit atteindre l’œil ou l’appareil récepteur, de sorte que les structures fluorescentes observées se superposent sur un fond très contrasté et très sombre (ou noir). L’obscurité de l’arrière-plan détermine généralement les limites de détection, puisque la lumière d’excitation est généralement des centaines de milliers, voire des millions de fois plus brillante que la fluorescence émise.La figure 1 montre une coupe schématique d'un microscope à épifluorescence moderne, conçu pour les observations en lumière transmise et réfléchie. L'éclairage vertical, situé au centre, comporte une source lumineuse à une extrémité (indiquée dans le schéma comme un module épiscopique) et un support de filtre à l'autre. La conception est basée sur un microscope fonctionnant en lumière réfléchie dont la longueur d’onde est supérieure à la longueur d’onde d’excitation. L'auteur de l'illuminateur vertical pour la microscopie à fluorescence en lumière réfléchie est considéré comme étant Johan S. Ploem. La lumière multifréquence provenant d'une lampe à arc ou d'une autre source, passant à travers un filtre d'excitation sélectif, est convertie dans un éclairage vertical fluorescent en lumière avec une certaine longueur d'onde (ou dans un intervalle d'onde donné), généralement à partir des parties ultraviolettes, bleues ou vertes. du spectre. Le flux traversant le filtre d'excitation est réfléchi par la surface d'un miroir dichromatique (également appelé dichroïque) ou d'un séparateur de faisceau et, en passant à travers l'objectif, illumine l'échantillon avec une lumière intense. Si l'échantillon est fluorescent, la lumière émise, collectée par l'objectif, traverse à nouveau le miroir dichroïque et est ensuite filtrée par un filtre de blocage (ou d'émission), qui bloque la lumière aux longueurs d'onde d'excitation. Il est important de noter que la fluorescence est le seul mode en microscopie optique dans lequel l’échantillon émet sa propre lumière après irradiation. La lumière émise est réémise de manière sphérique dans toutes les directions, quel que soit l'emplacement de la source lumineuse irradiante.
L’éclairage par épifluorescence est la méthode prédominante en microscopie moderne. L'illuminateur vertical à lumière réfléchie est situé entre les tubes d'observation et la tête tournante des lentilles. L'illuminateur est conçu de telle manière que la lumière d'excitation en direction de l'image et en provenance de l'échantillon traverse le même objectif du microscope, qui dans cette configuration agit d'abord comme un condenseur, et collecte sur le chemin de retour la lumière de fluorescence émise. Les briquets de ce type présentent plusieurs avantages. La lentille d'un microscope à fluorescence agit, d'une part, comme un condenseur bien réglé, et, d'autre part, comme un dispositif de collecte de lumière avec lequel une image est formée. Étant le même composant, l’objectif/condensateur est toujours parfaitement aligné. La majeure partie de la lumière d'excitation atteignant l'échantillon le traverse sans interaction et ne retourne pas vers l'objectif, et la zone éclairée est limitée à la partie de l'échantillon observée à travers les oculaires (dans la plupart des cas). Si le microscope est correctement configuré pour l'éclairage de Köller, contrairement à certaines techniques d'amélioration du contraste, la pleine ouverture numérique de l'objectif est disponible pour la visualisation. De plus, il permet de combiner les modes d'observation en lumière transmise et réfléchie, le mode de formation d'image numérique, ou d'en sélectionner un.
Riz. 2. Filtres fluorescents
Comme le montre la figure 1, à l'extrémité arrière de l'illuminateur à lumière réfléchie verticale se trouve une lampe à arc (généralement au mercure ou au xénon). Se propageant le long de l'illuminateur perpendiculairement à l'axe optique du microscope, la lumière d'excitation traverse les lentilles collectrices, le diaphragme à ouverture réglable et centrable, puis à travers le diaphragme de champ réglable et centrable (voir Figure 1). Après cela, la lumière pénètre dans le filtre d'excitation, où les longueurs d'onde sont sélectionnées dans l'intervalle requis et les longueurs d'onde restantes sont bloquées. Après avoir traversé le filtre d'excitation, les longueurs d'onde sélectionnées atteignent le miroir de séparation de faisceau dichroïque, qui est un filtre d'interférence spécial qui reflète efficacement la lumière de courte longueur d'onde et transmet efficacement la lumière de longue longueur d'onde. Le séparateur de faisceau dichroïque est incliné à un angle de 45 degrés par rapport à la lumière d'excitation incidente et la réfléchit à un angle de 90 degrés à travers la lentille du système optique directement sur l'échantillon. La fluorescence émise par l'échantillon éclairé est captée par la lentille, qui remplit désormais sa fonction habituelle, à savoir la formation d'images. Étant donné que les longueurs d'onde émises sont plus longues que les longueurs d'onde d'excitation, elles traversent le miroir dichroïque vers le haut jusqu'aux tubes d'observation ou au détecteur d'électrons.La majeure partie de la lumière d’excitation diffusée atteignant le miroir dichroïque est réfléchie vers la source lumineuse, bien qu’une petite fraction puisse traverser ou être absorbée par le revêtement interne du miroir. Mais avant que la fluorescence émise n'atteigne l'oculaire ou le détecteur, elle doit passer à travers un filtre de blocage ou de rejet. Ces filtres bloquent (bloquent) toute lumière d'excitation résiduelle, mais laissent passer des longueurs d'onde plus longues de lumière émise. La plupart des illuminateurs à lumière réfléchie combinent le filtre d'excitation, le miroir dichroïque et le filtre d'arrêt dans une unité optique (souvent appelée cube), comme le montre la figure 2. Les microscopes à fluorescence modernes peuvent accueillir quatre à six cubes filtrants (généralement sur un type carrousel ou tiroir). (accessoire) ; voir Figure 1) et permettent à l'utilisateur d'installer facilement des filtres d'excitation, des filtres de blocage et des miroirs dichroïques remplaçables.
La conception de l'illuminateur vertical doit permettre à l'utilisateur d'ajuster le microscope pour l'éclairage de Köller, qui fournit un éclairage brillant et uniforme sur tout le champ de vision. Les lentilles à condensateur corrigées du système optique garantissent que l'image du diaphragme à ouverture centrée est alignée avec l'ouverture arrière de l'objectif de mise au point. Dans les éclairages modernes, l'image d'un diaphragme à champ centré et préfocalisé est conjuguée à l'image focalisée et au plan du diaphragme fixe de l'oculaire.
L'unité de lampe éclairante contient généralement un filtre d'arrêt qui bloque la lumière infrarouge. La lampe elle-même ne doit pas transmettre de rayonnement ultraviolet vers l’extérieur. Il est également souhaitable qu'il dispose d'un disjoncteur intégré au cas où il s'ouvrirait pendant le fonctionnement. La lampe doit être suffisamment solide pour résister à une éventuelle explosion de la lampe à arc pendant le fonctionnement. Dans les lampes modernes, la douille de la lampe est équipée de boutons de réglage pour centrer l'image de la lampe à arc dans l'ouverture arrière de l'objectif (sous éclairage Keller, ces plans sont conjugués). Il est conseillé de placer un obturateur sur le trajet de la lumière, généralement à proximité de la lampe mais avant le filtre d'excitation, pour bloquer complètement la lumière d'excitation à moins que l'échantillon ne soit observé. De plus, l'équipement de l'illuminateur doit comprendre des filtres à densité neutre (sur fixation de type tambour, carrousel ou rétractable) afin de pouvoir réduire l'intensité de l'éclairage d'excitation.
Changement de Stokes
Lorsque les électrons passent de l’état excité à l’état fondamental, l’énergie vibratoire est perdue. En raison de cette perte d'énergie, le spectre d'émission du fluorophore excité se déplace généralement vers des longueurs d'onde plus longues par rapport au spectre d'absorption ou d'excitation (rappelez-vous que la longueur d'onde est inversement proportionnelle à son énergie). Ce phénomène bien connu est appelé règle de Stokes ou décalage de Stokes. À mesure que le décalage de Stokes augmente, il devient plus facile de séparer la lumière d’excitation et d’émission à l’aide de combinaisons de filtres fluorescents.L'intensité maximale d'émission d'un fluorophore est généralement inférieure à l'intensité maximale de son absorption et se produit à une longueur d'onde plus longue. La courbe d'émission (courbe spectrale) est souvent une image miroir (ou proche) de la courbe d'excitation, mais décalée vers des longueurs d'onde plus longues, comme le montre la figure 3, qui montre les caractéristiques spectrales utiles du colorant Alexa Fluor 555, qui absorbe dans le jaune-vert et émet dans la région jaune-orange. Pour obtenir une intensité de fluorescence maximale, le fluorophore (souvent appelé colorant) est excité à des longueurs d'onde proches ou proches du pic de la courbe d'excitation, et la lumière émise est détectée sur la plage la plus large possible incluant le pic d'émission. La sélection des longueurs d'onde excitatrices et émises est réalisée à l'aide de filtres interférentiels (Figure 2). De plus, il convient de noter que les caractéristiques spectrales du système optique du microscope dépendent également de la transmission du verre (qui est affectée par les revêtements antireflet), du nombre de lentilles et de miroirs et de la sensibilité des détecteurs.
Riz. 3. Courbes d'absorption et d'émission des fluorophores
L'efficacité de la séparation et de l'enregistrement des longueurs d'onde d'excitation et d'émission est obtenue en microscopie à fluorescence grâce au choix correct de filtres de lumière qui bloquent ou, au contraire, transmettent la lumière de certaines longueurs d'onde dans les régions ultraviolettes, visibles et proches infrarouges du spectre. La lumière d'excitation est contrôlée dans les éclairages fluorescents verticaux du fait que leur conception prévoit l'utilisation de filtres facilement remplaçables (filtres d'excitation neutres et interférentiels) insérés le long du trajet lumineux vers l'échantillon et sur le trajet de retour entre l'échantillon et les tubes d'observation. ou système de réception de signal. En raison de la faible intensité de l'émission fluorescente (comme indiqué ci-dessus), il est nécessaire que la source de lumière d'excitation ait une luminosité suffisante pour maximiser la faible lumière émise, et que les fluorochromes aient des caractéristiques d'absorption et une efficacité quantique appropriées. C’est peut-être le critère clé de la microscopie à fluorescence.
L'efficacité avec laquelle un fluorophore individuel absorbe un photon de lumière d'excitation est fonction de sa section efficace moléculaire, et la probabilité qu'un tel événement se produise est appelée coefficient d'absorption. Des valeurs plus grandes du coefficient d'absorption indiquent que l'absorption d'un photon (ou d'un quantum) dans un intervalle de longueur d'onde donné est plus probable. Le rendement quantique est le rapport entre le nombre de quanta émis et le nombre de quanta absorbés (il se situe généralement entre 0,1 et 1,0). Le fait que le rendement quantique prenne des valeurs inférieures à 1 est une conséquence d'une perte d'énergie de manière non radiative, par exemple par la chaleur ou une réaction photochimique, lorsque la réémission ne se produit pas, conduisant à la fluorescence. Le coefficient d'absorption, le rendement quantique, l'intensité lumineuse moyenne et le temps d'exposition sont des facteurs importants affectant l'intensité de fluorescence et déterminant la faisabilité de l'utilisation de cette méthode.
Décoloration, décoloration et photoblanchiment
Un certain nombre de conditions peuvent affecter la probabilité de réémission de fluorescence, entraînant souvent une baisse de l'intensité de la fluorescence. Le terme général désignant une diminution de l'intensité de l'émission fluorescente est l'évanouissement, couvrant tous les phénomènes qui, pour une description plus détaillée, peuvent être divisés en phénomènes d'extinction et de photoblanchiment. Le photoblanchiment est la dégradation irréversible de molécules fluorescentes dans un état excité, provoquée par leur interaction avec l'oxygène moléculaire avant leur émission. Ce phénomène est utilisé dans la méthode de récupération de fluorescence après photoblanchiment (FRAP), très efficace pour étudier les propriétés de diffusion et le mouvement des macromolécules biologiques. La méthode est basée sur le photoblanchiment avec un faisceau laser d’une zone clairement définie de l’échantillon, suivi de l’observation du taux et de la nature de la récupération de fluorescence dans la zone photoblanchie. Une technique connexe, la décroissance de la fluorescence dans les images blanchies (FLIP), est utilisée pour étudier la diminution de la fluorescence dans les régions adjacentes à la région photoblanchie. Comme FRAP, cette méthode est un outil efficace pour étudier la mobilité et la dynamique moléculaire des cellules vivantes.Riz. 4. Taux de photoblanchiment d’échantillons multicolores
La figure 4 montre un exemple typique de photoblanchiment observé dans une série d’images numériques d’une culture multicolorée de fibroblastes cutanés de muntjac indien prises à différents moments. Les noyaux ont été colorés avec un dérivé de bis-benzimidazole (Hoechst 33258, fluorescence bleue), et les mitochondries et le cytosquelette d'actine ont été colorés avec MitoTracker Red CMXRos (fluorescence rouge) et un dérivé de phalloïdine couplé à Alexa Fluor 488 (fluorescence verte), respectivement. Des images ont été prises toutes les deux minutes et la combinaison de filtres fluorescents a été ajustée de manière à ce que les trois fluorophores soient excités simultanément, tout en enregistrant simultanément les signaux émis combinés. La figure 4 (a) montre que l'intensité des trois fluorophores est relativement élevée, mais que l'intensité de Hoechst (bleu) commence à diminuer rapidement après seulement deux minutes et disparaît presque complètement au bout de 6 à 8 minutes. Les colorants mitochondriaux et actines semblent plus résistants au photoblanchiment, mais leur intensité diminue également de manière significative au cours de la période d'observation (10 minutes).La relaxation de l'état excité par trempe, entraînant une baisse de l'intensité de la fluorescence, se produit de diverses manières non radiatives et est souvent due à des agents oxydants ou à la présence de sels, de métaux lourds et de composés halogènes. Dans certains cas, l'extinction se produit à la suite du transfert d'énergie vers une autre molécule (appelée accepteur) proche du fluorophore excité (donneur). Ce phénomène est appelé transfert d'énergie par résonance de fluorescence (FRET). C'est ce mécanisme qui est devenu la base d'une méthode efficace pour étudier les interactions et associations moléculaires à des distances nettement inférieures à la résolution des microscopes optiques.
Sources lumineuses fluorescentes
Une conséquence malheureuse de la faible intensité d’émission dans la plupart des applications de microscopie à fluorescence est le faible nombre de photons atteignant l’oculaire ou le récepteur. Dans la plupart des cas, l'efficacité de la collecte de photons dans les microscopes optiques est inférieure à 30 pour cent et les concentrations de nombreux fluorophores le long du trajet optique vont de concentrations micromolaires à nanomolaires. Pour que l’intensité lumineuse d’excitation soit suffisante pour détecter la fluorescence, de puissantes sources lumineuses compactes, telles que de petites lampes à arc à haute énergie, sont nécessaires. Les plus courantes sont les lampes au mercure d'une puissance de 50 à 200 watts et les lampes au xénon d'une puissance de 75 à 150 watts (voir figure 5). Ces lampes sont généralement alimentées par une source CC externe suffisante pour allumer l'arc par ionisation de vapeur à haute pression et le maintenir allumé avec un scintillement minimal.L'alimentation de la lampe à arc externe du microscope est généralement équipée d'une minuterie pour suivre le nombre d'heures d'utilisation. Les lampes à arc perdent leur puissance lumineuse et tombent souvent en panne lorsqu'elles sont utilisées au-delà de leur durée de vie spécifiée (200 à 300 heures). Les lampes au mercure ne fournissent pas une intensité uniforme sur toute la plage spectrale allant des UV aux IR. Leur intensité maximale se produit dans le proche ultraviolet. Des pics d'intensité distincts se produisent à 313, 334, 365, 406, 435, 546 et 578 nanomètres. Aux autres longueurs d’onde du spectre visible, l’intensité est stable, bien que moins élevée (mais néanmoins suffisante pour la plupart des applications). Mais la puissance de la lampe elle-même n’est pas un paramètre déterminant pour l’efficacité de l’éclairage. A l'inverse, un paramètre essentiel qu'il faut en premier prendre en compte est la luminosité moyenne, prenant en compte la luminosité de la source, la géométrie de l'arc et la répartition angulaire du rayonnement.
Riz. 5. Lampes fluorescentes à arc
Au cours des dernières années, la microscopie optique a connu une recrudescence de l'utilisation de sources de lumière laser, en particulier les lasers à ions argon et argon-krypton (ions). Les avantages de ces lasers sont leur petite taille, leur faible divergence de faisceau, leur degré élevé de monochromaticité et leur luminosité moyenne élevée. Ils sont largement utilisés en microscopie confocale à balayage, qui est devenue un outil puissant pour créer des images fluorescentes à contraste élevé en éliminant la lumière floue provenant du plan focal de l'échantillon. Dans les microscopes confocaux, ceci est réalisé en balayant l'échantillon avec un point focal ou une ligne tout en formant simultanément une image à travers une ouverture conjuguée. Les sections optiques des échantillons peuvent être stockées dans la mémoire de l'ordinateur du microscope et reconstruites en une image finale affichée sur un moniteur.Symboles de filtre
La terminologie générale adoptée pour désigner les combinaisons de filtres en microscopie à fluorescence est devenue assez confuse en raison des différentes abréviations et codes utilisés par les différents fabricants pour étiqueter leurs filtres. En principe, il existe trois grandes catégories de filtres : les filtres d'excitation (souvent simplement appelés excitateurs), les filtres de blocage (d'émission) et les séparateurs de faisceaux dichroïques (ou miroirs dichroïques). Auparavant, les filtres fluorescents étaient constitués uniquement de verre coloré ou de gélatine pris en sandwich entre deux plaques de verre. Cependant, il existe aujourd'hui une tendance à produire des filtres très sensibles avec une optique interférentielle pour transmettre ou retarder la lumière de longueurs d'onde strictement définies, qui ont également une transmission élevée. Les séparateurs de faisceau dichroïque sont des filtres interférentiels spéciaux conçus pour réfléchir ou transmettre la lumière de longueurs d'onde spécifiques lorsqu'ils sont placés à un angle de 45 degrés le long du trajet lumineux (voir figures 1 et 2). Les filtres bloquants sont fabriqués à partir de verre coloré ou de revêtements interférentiels (ou une combinaison des deux).Les fabricants utilisent différentes abréviations pour indiquer les caractéristiques des filtres d'excitation. Le verre ultraviolet, par exemple, est désigné UG et le verre bleu, BG. Sur les filtres à bande étroite, vous pouvez souvent voir la désignation KP (K de l'allemand « kurz », qui se traduit par « court ») ou simplement SP. Les filtres antiparasitaires sont désormais étiquetés par certains fabricants avec l'abréviation IF. Les filtres d'excitation interférentielle à bande étroite sont particulièrement efficaces pour les faibles déplacements de Stokes.
Les abréviations et abréviations des filtres bloquants sont les suivantes : LP ou L pour les filtres à large bande, Y ou GG pour le verre jaune (de l'allemand "gelb" - jaune), R ou RG pour le verre rouge, OG ou O pour le verre orange, K pour filtres à fentes (de l'allemand « kante » - bord) et BA pour bloquer les filtres. Si un filtre porte un numéro, tel que BA515, il indique la longueur d'onde (en nanomètres) à laquelle il a la moitié de sa transmission maximale.
Les séparateurs de faisceau dichroïque portent également diverses abréviations : CBS signifie séparateur de faisceau chromatique, DM signifie miroir dichroïque, TK signifie séparateur de fentes (de l'allemand "teiler kante"), FT signifie séparateur de couleurs (de l'allemand "farb teiler"). "), et RKP signifie réflecteur à bande étroite. Toutes ces désignations sont interchangeables. De plus, le verre optique de tous les séparateurs de faisceau dichroïque modernes est toujours recouvert de revêtements interférentiels (plutôt que de colorants organiques ou métalliques). haute réflectance des courtes longueurs d'onde et haute transmission des longues longueurs d'onde. Les séparateurs de faisceau dichroïque sont inclinés à un angle de 45 degrés par rapport à la lumière d'excitation incidente sur le bloc optique à travers l'illuminateur fluorescent de la lumière réfléchie. plus courtes) à la lentille et à l'échantillon situé derrière celle-ci. Ces filtres spéciaux ont également pour fonction supplémentaire de transmettre des ondes de fluorescence plus longues au filtre bloquant et de réfléchir la lumière d'excitation diffusée vers la lampe.
Riz. 6. Filtre d'excitation bleu mi-bande Nikon B-2E
La figure 6 montre les courbes de transmission pour une combinaison de filtres fluorescents typiques utilisés dans les microscopes modernes. Le spectre du filtre d'excitation (courbe rouge) présente une transmission élevée (environ 75 %) dans la plage de 450 à 490 nanomètres avec une longueur d'onde centrale (CWL) de 470 nanomètres. Le miroir dichroïque (courbe jaune) réfléchit les ondes dans le domaine spectral du filtre d'excitation, mais transmet, avec un coefficient relativement élevé, des ondes de plus en plus courtes. Il convient de noter que la transmission nulle d'un miroir dichroïque correspond à une réflexion de 100 pour cent. Le creux distinct de la courbe de transmission entre 450 et 500 nanomètres, qui correspond au pic de réflexion, sert à rediriger les ondes de la bande passante du filtre d'excitation à 90 degrés vers l'échantillon. Le dernier maillon de cette séquence est le filtre d'émission ou de blocage (courbe blanche), qui transmet des ondes dans la partie verte du spectre visible dans la plage de 520 à 560 nanomètres. Pour assurer une séparation quasi complète des ondes réfléchies et transmises, les limites des bandes de réflexion et de transmission des différents spectres superposés doivent être aussi raides que possible. La partie sinusoïdale de la courbe spectrale du miroir dichroïque, appelée sonnerie, est le résultat du processus de dépôt de couches minces. La haute efficacité de cette combinaison de filtres est un exemple d’avancées significatives dans la technologie des filtres interférentiels à couche mince.Les conventions de dénomination de Nikon sont basées sur des termes mixtes apparus au début des années 1990. À cette époque, toutes les combinaisons de filtres supplémentaires de Nikon étaient produites à l'aide de la méthode de revêtement dur, mais aujourd'hui, de nombreux filtres sont fabriqués à l'aide de méthodes avancées de revêtement doux. Bien que les revêtements souples soient plus sensibles à l’humidité et à la chaleur et nécessitent une manipulation plus soigneuse (par rapport aux revêtements durs), ils présentent des densités optiques plus élevées et facilitent davantage le réglage précis de bandes passantes spécifiques. Comprendre les conventions des combinaisons de filtres Nikon vous permet de sélectionner rapidement les filtres nécessaires pour des fluorophores spécifiques.
La première lettre du système de notation alphanumérique de Nikon indique la région du spectre d'excitation (par exemple, UV, V, B et G sont les abréviations des mots anglais "ultraviolet" - ultraviolet, "violet" - violet, "blue" et " vert” - vert, respectivement). Le chiffre suivant le codage du spectre d'excitation indique la bande passante du filtre d'excitation : 1 correspond à une excitation bande étroite, 2 à une excitation bande moyenne et 3 à une excitation large bande. Enfin, une ou plusieurs lettres suivant le chiffre correspondant à la bande passante d'excitation indiquent les caractéristiques du filtre bloquant. La lettre A indique le filtre de coupure large bande standard avec la longueur d'onde de coupure la plus basse, B indique le filtre de coupure large bande ayant une longueur d'onde de coupure plus élevée. La désignation E (de l'anglais « Enhanced » - Enhanced) dans les filtres d'émission passe-bande indique des performances améliorées dans le sens de réduction de l'interaction interférentielle des signaux séparés. La désignation E/C indique une combinaison de revêtements interférentiels doux conçus spécifiquement pour fonctionner avec des colorants spécifiques tels que le DAPI, le FITC, le TRITC et le Texas Red.
Balance de lumière fluorescente
L'évaluation des flux lumineux d'un microscope à fluorescence typique donne une idée générale des limitations qui surgiront inévitablement lors de la génération d'images numériques ou de l'observation visuelle d'échantillons. La source d'irradiation pour notre évaluation sera une lampe à arc au xénon standard de 75 watts, qui a une densité de flux lumineux moyenne d'environ 400 candelas par millimètre carré (d'autres sources sont présentées dans le tableau 1). Lorsque la lumière émise est dirigée vers un filtre interférentiel de 490 nanomètres (avec une bande passante de 10 nanomètres et une transmission de 75 %), environ 2 milliwatts de puissance de la lampe le traversent. Après réflexion sur un miroir dichroïque avec un coefficient de 0,9, un flux lumineux de 1,8 milliwatts est dirigé vers l'ouverture arrière de l'objectif du microscope sous forme de faisceau d'excitation.Pour un objectif 100x avec une ouverture numérique de 1,4, la zone éclairée de l'échantillon sera de 12 x 10 E(-6) centimètres carrés, si le diamètre du champ de vision est supposé être d'environ 40 micromètres. Le flux lumineux incident sur l’échantillon sera alors d’environ 150 watts par centimètre carré, ce qui correspond à une densité de flux de 3,6 × 10 E (20) photons par centimètre carré. Ainsi, l’intensité d’éclairage de l’échantillon est environ 1 000 fois supérieure à l’intensité d’éclairage de la surface terrestre lors d’une journée ensoleillée normale.
L'émission fluorescente à un tel flux lumineux dépend des propriétés d'absorption et d'émission du fluorophore, de sa concentration dans l'échantillon et de la longueur du trajet optique de l'échantillon. La fluorescence (F) produite mathématiquement est décrite par l'équation :
F = σ Q je
Où σ est la section efficace d'absorption moléculaire, Q est le rendement quantique et I est le flux lumineux incident calculé ci-dessus. En supposant que la fluorescéine est un fluorophore avec une section efficace d’absorption (σ) de 3 × 10 E(-16) centimètres carrés, nous obtenons un Q de 0,99, ce qui se traduit par une fluorescence F de 100 000 photons par seconde et par molécule. À une concentration de colorant de 1 micromole par litre, uniformément répartie dans un disque de 40 micromètres de diamètre et 10 micromètres d'épaisseur (volume égal à 12 picolitres), cela donne environ 1,2 x 10 E(-17) moles de colorant, soit 7,2 millions molécules dans le chemin optique. Si toutes les molécules sont excitées simultanément, le taux de fluorescence sera de 7,2 x 10 photons E(11) par seconde (qui est le produit de F et du nombre de molécules de colorant). La question se pose : combien de photons émis seront enregistrés, et combien de temps ce taux d’émission peut-il perdurer ?
Tableau 1. Densité d'énergie lumineuse de diverses sources lumineuses
L'efficacité de la détection des photons est déterminée par l'efficacité de leur collecte et le rendement quantique du détecteur. Un objectif avec une ouverture numérique de 1,4 et une transmission de 100 pour cent (ce qui est une condition irréaliste) a une efficacité maximale de collecte de photons limitée à un angle d'acceptation d'environ 30 pour cent. La transmission du miroir dichroïque est de 85 pour cent et celle du filtre bloquant est de 80 pour cent. L’efficacité de collecte qui en résulte dans ce cas est de 20 %, soit 140 milliards de photons par seconde. Si un dispositif à couplage de charge (CCD) traditionnel est utilisé comme détecteur, son rendement quantique à la longueur d'onde de la fluorescéine verte (525 nanomètres) sera de 50 %. Ainsi, 70 milliards de photons par seconde seront détectés, soit environ 10 % de ceux émis par fluorescence. Même un détecteur idéal (avec une efficacité quantique de 100 %) ne peut capturer qu’environ 20 % des photons de fluorescence.
La durée de la lueur fluorescente dépend du taux de destruction des fluorophores, conséquence du photoblanchiment. Les mesures montrent que chaque molécule de fluorescéine présente dans une solution saline contenant de l'oxygène parvient à émettre environ 36 000 photons avant d'être détruite. Dans un environnement sans oxygène, le taux de photodestruction est réduit d’environ dix fois. Ainsi, une molécule de fluorescéine peut produire 360 000 photons. Collectivement, tous les colorants de notre exemple (7,2 millions de molécules) sont capables d’émettre un minimum de 2,6 x 10 E(11) et un maximum de 2,6 x 10 E(12) photons. A un taux d'émission d'une molécule de 100 000 photons par seconde (d'après les estimations ci-dessus), on obtient la durée d'émission fluorescente avant photodestruction égale à 0,3 à 3 secondes. Si 10 pour cent des photons émis sont détectés, le signal du détecteur sera de 7,2 × 10 E(10) électrons par seconde.
Ainsi, si un CCD dispose d'une caméra de 1 000 x 1 000 pixels, ce signal sera réparti entre un million d'éléments photosensibles, soit environ 72 000 électrons pour chacun d'eux. Pour un CCD de recherche doté d'éléments photosensibles de 9 micromètres, la capacité de charge est d'environ 80 000 électrons et le bruit de lecture est inférieur à 10 électrons. Dans ce cas, le rapport signal sur bruit sera principalement déterminé par un bruit de fluctuation de photons égal à la racine carrée du signal, soit environ 268. Dans presque tous les cas, un niveau de signal aussi élevé ne peut durer qu'une courte période. temps avant que la photodestruction ne se produise. Pour prolonger la durée d'observation, la plupart des microscopistes réduisent l'intensité du flux irradiant de sorte que seule une partie du nombre total de molécules fluorophores soit excitée, et donc détruite. Ainsi, le rapport signal sur bruit atteint rarement le maximum théorique et se situe généralement entre 10 et 20 en microscopie à fluorescence.
Détection d'une seule molécule
Dans des conditions idéales, il est souvent possible de détecter l’émission fluorescente d’une seule molécule, à condition, bien entendu, que le fond optique et le bruit du détecteur soient suffisamment faibles. Comme mentionné ci-dessus, une molécule de fluorescéine peut émettre jusqu'à 300 000 photons avant d'être détruite par photoblanchiment. Avec une efficacité de collecte et de détection de 20 %, environ 60 000 photons seront détectés. En utilisant des CCD basés sur des photodiodes à avalanche ou sur la multiplication d'électrons pour des expériences de ce type, les chercheurs ont pu surveiller le comportement de molécules individuelles sur une période de quelques secondes, voire minutes. Le principal problème dans de tels cas est la suppression du bruit de fond optique. Étant donné que de nombreux matériaux utilisés dans la construction de lentilles et de filtres microscopiques présentent un certain degré d’autofluorescence, les efforts initiaux ont été orientés vers la production de composants à faible fluorescence. Cependant, il est vite devenu évident qu’en utilisant la réflexion interne totale (TIR) en microscopie à fluorescence, la combinaison requise d’une lumière de fond faible et d’une lumière d’excitation de haute intensité pouvait être obtenue.
Riz. 7. Configurations de microscopes inversés et TIRF
La microscopie à fluorescence par réflexion interne totale (TIRFM ou TIRFM dans son acronyme anglais) utilise le phénomène d'onde non propagée ou décroissante rapidement qui se produit lors de la réflexion interne totale à l'interface de deux milieux d'indices de réfraction différents.
Un circuit utilisant une source de lumière externe est illustré à la figure 7 (a). Dans cette méthode, un faisceau de lumière (généralement un faisceau laser étendu) traverse un prisme à indice de réfraction élevé (comme du verre ou du saphir) adjacent au verre ou à une solution aqueuse à indice de réfraction inférieur. Si la lumière est dirigée vers un prisme selon un angle supérieur à l’angle critique, le faisceau sera complètement réfléchi par l’interface. Le phénomène de réflexion provoque une onde non propagée à l'interface, c'est-à-dire qu'un champ électromagnétique est généré qui pénètre dans un milieu avec un indice de réfraction inférieur jusqu'à une distance ne dépassant pas 200 nanomètres. L'intensité lumineuse de l'onde évanescente est suffisante pour exciter les fluorophores, mais en raison de sa profondeur extrêmement faible, la quantité d'excitation est très faible. Il en résulte un bruit de fond de faible niveau, car le volume de l'échantillon exposé à l'irradiation est négligeable (uniquement la partie située à moins de 200 nm de la surface).
La microscopie à fluorescence par réflexion interne totale peut également être réalisée à l'aide d'une technique d'épi-illumination modifiée utilisée en microscopie à grand champ (comme le montre la figure 7 (b)). Cette méthode nécessite des objectifs à NA très élevés (au moins 1,4, mais de préférence 1,45 à 1,6) et un champ de microscope partiellement éclairé, obtenu en utilisant un petit point ou, pour une plus grande uniformité d'éclairage, un anneau fin qui bloque une partie de la lumière. flux. Pour atteindre l'angle critique au-delà duquel la réflexion interne totale se produit, il est nécessaire que le milieu d'immersion dans les lentilles et le verre de protection du microscope ait un indice de réfraction élevé. Comme le montre la figure 7 (b), les rayons lumineux quittant la lentille frontale sous un angle inférieur à l'angle critique (sur la figure, il est désigné A (1)) ne reviennent pas au microscope. Lorsque l'angle critique est atteint ou dépassé (angle A(2) sur la figure 7(b)), une réflexion interne totale se produit.
Pour fournir des informations supplémentaires dans la recherche, la réflexion interne totale est souvent combinée à d'autres techniques avancées de fluorescence populaires telles que le transfert d'énergie par résonance de fluorescence (FRET), la récupération de fluorescence après photoblanchiment (FRAP) et la spectroscopie. En combinaison, ces méthodes constituent des outils puissants dans l’étude de fluorophores individuels et de molécules fluorescentes. Les avantages de l’étude de molécules uniques commencent seulement à se faire sentir. Ainsi, aujourd’hui, l’éventail des études en microscopie optique s’étend des molécules uniques aux animaux entiers.
Conclusion
Les microscopes à fluorescence modernes combinent la puissance de composants optiques de haute qualité avec un contrôle informatisé et une imagerie numérique pour atteindre un niveau de sophistication qui dépasse de loin la simple observation visuelle. Aujourd'hui, la microscopie s'appuie fortement sur les techniques d'imagerie électronique pour obtenir rapidement des informations à de faibles niveaux de signaux lumineux ou à des longueurs d'onde visuellement indétectables. Ces améliorations techniques ne sont pas seulement des éléments de conception externe, mais aussi des composants essentiels du microscope optique en tant que système de mesure complexe.
Le temps où la microscopie optique était une discipline purement descriptive ou un jouet intellectuel est révolu. Aujourd’hui, l’imagerie optique n’est que la première étape de l’analyse des données. Cette première étape est réalisée par un microscope en association avec des détecteurs électroniques, des processeurs d'images, des écrans, qui peuvent être considérés comme une extension du système d'imagerie. Le contrôle informatisé de la mise au point, de la position de la platine, des composants optiques, des obturateurs, des filtres et des détecteurs, déjà couramment utilisé, permet de telles manipulations lors d'expériences qui étaient tout simplement impossibles pour les humains travaillant sur des microscopes mécaniques. L'utilisation croissante de l'optoélectronique en microscopie à fluorescence a conduit au développement de pinces optiques pour la manipulation de structures et de particules subcellulaires, l'observation de molécules individuelles et une large gamme d'applications spectroscopiques sophistiquées.
Combinaisons de filtres fluorescents
Des combinaisons de filtres d'interférence et d'absorption d'épifluorescence sont placées dans des cubes filtrants (ou unités optiques) et comprennent un filtre d'excitation, un séparateur de faisceau dichroïque (souvent appelé miroir) et un filtre de blocage (ou d'émission), comme le montre la figure 1. (un). Ce guide peut être utile pour sélectionner des combinaisons de filtres qui correspondent aux caractéristiques spectrales d’absorption et d’émission des chromophores utilisés en microscopie à fluorescence à grand champ. Les courbes spectrales d'une combinaison typique de filtres passe-bande hautes performances dans la plage d'excitation bleue sont illustrées à la figure 1 (b). Les combinaisons de filtres de fluorescence Nikon sont disponibles avec des filtres d'excitation à bande étroite, moyenne et large bande et leurs filtres d'émission spécifiques ou à large bande correspondants.
Riz. 8. Courbes spectrales d'un bloc de filtres de lumière fluorescente
Excitation ultraviolette - L'ensemble de filtres de fluorescence d'excitation UV de Nikon comprend quatre combinaisons soigneusement équilibrées qui incluent des filtres passe-bande conventionnels ou des filtres d'émission à large bande (blocage) qui peuvent transmettre sélectivement l'émission fluorescente dans une plage étroite ou large des parties bleues, vertes et rouges du spectre visible. Ces combinaisons de filtres couvrent la plage d'excitation de 330 à 380 nanomètres avec des bandes passantes de 10, 40 et 50 nanomètres. Trois combinaisons utilisent le même miroir dichroïque et la quatrième a une longueur d'onde de coupure inférieure pour correspondre à la bande d'excitation étroite. Les combinaisons de filtres UV comprennent des filtres d'émission à bande passante fixe ou à large bande.
Excitation violette : le jeu de filtres fluorescents à excitation violette de Nikon comprend trois combinaisons comprenant des filtres passe-bande ou d'émission à large bande (blocage) conventionnels qui peuvent transmettre sélectivement l'émission fluorescente dans une plage étroite ou large de régions bleues, vertes et rouges du spectre. Ces combinaisons de filtres couvrent la plage d'excitation de 379 à 420 nanomètres avec des bandes passantes de 10, 22 et 40 nanomètres. Deux combinaisons utilisent le même miroir dichroïque et la troisième a une longueur d'onde de coupure inférieure pour correspondre à la bande d'excitation aux longueurs d'onde plus courtes.
Excitation bleu-violet - L'ensemble de filtres de fluorescence d'excitation bleu-violet de Nikon comprend quatre combinaisons comprenant des filtres passe-bande conventionnels ou des filtres d'émission à large bande (blocage) qui peuvent transmettre sélectivement l'émission fluorescente dans une plage étroite ou large des régions bleues, vertes et rouges du visible. spectre. Ces jeux de filtres supplémentaires couvrent la plage d'excitation de 400 à 446 nanomètres avec des bandes passantes de 10, 20 et 40 nanomètres. Trois combinaisons utilisent le même miroir dichroïque, tandis que la quatrième a une longueur d'onde de coupure plus élevée (de 5 nanomètres) pour correspondre aux autres composants.
Excitation bleue : le jeu de filtres fluorescents à excitation bleue de Nikon se compose de six combinaisons équilibrées comprenant des filtres passe-bande ou d'émission à large bande (blocage) conventionnels qui peuvent transmettre sélectivement l'émission fluorescente dans une plage étroite ou large des régions vertes, jaunes, rouges et infrarouges du spectre. . Ces combinaisons de filtres couvrent la plage d'excitation de 420 à 495 nanomètres avec des bandes passantes de 20, 30, 40 et 70 nanomètres. Cinq combinaisons utilisent le même miroir dichroïque et la sixième a une longueur d'onde de coupure inférieure pour augmenter le signal reçu. Tous les filtres coupe-bande pour les kits de filtration par excitation bleue Nikon ont une largeur spectrale de 40 nanomètres. L'un des filtres (B-3A) est conçu pour être utilisé avec un éclairage à lampe halogène au tungstène.
Excitation verte - Le jeu de filtres fluorescents à excitation verte de Nikon se compose de six blocs, qui comprennent des filtres passe-bande ou d'émission à large bande (blocage) conventionnels capables de transmettre sélectivement l'émission fluorescente dans une plage étroite ou large de régions jaunes, oranges, rouges et proches infrarouges du spectre. Ces combinaisons de filtres couvrent une plage d'excitation de 510 à 560 nanomètres avec des bandes passantes de 10, 25, 30 et 50 nanomètres (y compris des bandes d'excitation étroites, moyennes et larges). Trois combinaisons utilisent le même miroir dichroïque (565 nanomètres) et les trois autres ont une longueur d'onde de coupure plus longue (570 et 575 nanomètres). Deux des six kits de filtres d'excitation verts de Nikon comprennent des filtres coupe-bande de 60 et 75 nanomètres.
Excitation jaune - Le jeu de filtres fluorescents à excitation jaune de Nikon se compose de deux combinaisons équilibrées comprenant des filtres d'émission (blocage) avec une bande passante spécifique, capables de transmettre sélectivement l'émission fluorescente dans les régions orange et rouge du spectre. Ces combinaisons de filtres optionnelles couvrent la plage d'excitation de 532 à 587 nanomètres avec des bandes passantes de 40 et 55 nanomètres. Les deux combinaisons incluent le même miroir dichroïque (avec une coupure à 595 nanomètres). Les deux kits de filtration d'excitation jaune de Nikon comprennent des filtres coupe-bande de 60 et 75 nanomètres.
Excitation rouge - La combinaison de filtres fluorescents à excitation rouge de Nikon est représentée par une seule unité, qui comprend un filtre d'émission passe-bande (blocage) capable de transmettre sélectivement l'émission fluorescente dans les régions du rouge lointain et du proche infrarouge du spectre. Le centre de la bande passante du filtre bloquant est de 700 nanomètres et sa largeur est de 75 nanomètres (de 663 à 738 nanomètres). La large bande d'excitation de 60 nm, de 590 à 650 nm, couvre les longueurs d'onde orange et rouge. La combinaison Cy5 HYQ comprend un miroir dichroïque avec une coupure à 660 nanomètres, soit 10 nanomètres plus élevée que la coupure de la bande d'excitation.
Excitation de la protéine fluorescente jaune (YFP) - Pour YFP, Nikon a développé une combinaison équilibrée de haute qualité qui améliore les capacités de détection des protéines fluorescentes (fournies par trois kits de filtres de protéine fluorescente verte (GFP)) en utilisant des filtres pour les variantes de GFP à longueur d'onde plus longue ( YFP et EYFP). La banque de filtres YFP HYQ comprend des filtres d'excitation et d'émission (arrêt) avec des bandes passantes relativement étroites conçues spécifiquement pour correspondre aux caractéristiques spectrales de la protéine fluorescente jaune améliorée par YFP (YFP), permettant d'évaluer la fluorescence des dérivés de YFP séparément des autres protéines fluorescentes.
Excitation double bande - L'ensemble de filtres de fluorescence double bande de Nikon se compose de trois combinaisons soigneusement équilibrées de filtres double bande (filtres d'excitation et d'émission (coupure)) combinés en une seule unité qui transmet simultanément de manière sélective l'émission fluorescente de deux fluorophores. Chacune des unités de filtration est combinée de manière optimale avec une paire de fluorochromes spécifique, bien qu'elle puisse également fonctionner efficacement avec d'autres paires de colorants fluorescents ayant les mêmes profils spectraux d'absorption et d'émission. Grâce à une adaptation précise des bandes, avec des transitions abruptes de bande à bande entre les régions de réflexion et de transmission, les différents signaux d'excitation et d'émission sont séparés avec un minimum d'interférences superposées.
Excitation à trois bandes - Les filtres fluorescents à trois bandes Nikon sont représentés par deux blocs équilibrés, comprenant des filtres à trois bandes (filtres d'excitation et d'émission (blocage)), transmettant sélectivement l'émission fluorescente de trois fluorophores simultanément. Chacune des unités de filtration est combinée de manière optimale avec un ensemble spécifique de trois fluorochromes, bien qu'elle puisse également fonctionner efficacement avec d'autres combinaisons de colorants présentant les mêmes profils spectraux d'absorption et d'émission. Grâce à une adaptation précise des bandes, avec des transitions interbandes abruptes entre les régions de réflexion et de transmission, les différents signaux d'excitation et d'émission sont séparés avec une interférence minimale entre eux. Triples cubes.
Cubes HYQ - Les combinaisons de filtres de fluorescence HYQ de Nikon comportent quatre blocs de haute qualité soigneusement équilibrés, chacun intégrant des filtres d'émission passe-bande (blocage) pour transmettre sélectivement la fluorescence dans une plage limitée. Chaque désignation de filtre HYQ reflète le nom du fluorochrome pour lequel il a été conçu, mais dans ses plages d'excitation, chaque combinaison peut être utilisée pour observer différents fluorochromes avec des caractéristiques correspondantes.
Liste de base des unités de filtrage fluorescentes Nikon
Les conventions de dénomination de Nikon sont basées sur des termes mixtes apparus au début des années 1990. À cette époque, toutes les combinaisons de filtres supplémentaires de Nikon étaient produites à l'aide de la méthode de revêtement dur, mais aujourd'hui, de nombreux filtres sont fabriqués à l'aide de méthodes avancées de revêtement doux. Bien que les revêtements souples soient plus sensibles à l’humidité et à la chaleur et nécessitent une manipulation plus soigneuse (par rapport aux revêtements durs), ils présentent des densités optiques plus élevées et facilitent davantage le réglage précis de bandes passantes spécifiques. Comprendre les conventions des combinaisons de filtres Nikon vous permet de sélectionner rapidement les filtres nécessaires pour des fluorophores spécifiques.
La première lettre du système de notation alphanumérique de Nikon indique la région du spectre d'excitation (par exemple, UV, V, B et G sont des abréviations des mots anglais "ultraviolet" - ultraviolet, "violet" - violet, "blue" - bleu , et « vert » - vert, respectivement). Le chiffre suivant le codage du spectre d'excitation indique la bande passante du filtre d'excitation : 1 correspond à une excitation bande étroite, 2 à une excitation bande moyenne et 3 à une excitation large bande. Enfin, une ou plusieurs lettres suivant le chiffre correspondant à la bande passante d'excitation indiquent les caractéristiques du filtre bloquant. La lettre A indique un filtre de coupure à large bande standard avec la longueur d'onde de coupure la plus basse, B indique un filtre d'émission à large bande ayant une longueur d'onde de coupure plus élevée. La désignation E (de l'anglais « Enhanced » - Enhanced) dans les filtres d'émission passe-bande indique des performances améliorées dans le sens de réduction de l'interaction interférentielle des signaux séparés. La désignation E/C indique une combinaison de revêtements interférentiels doux conçus spécifiquement pour fonctionner avec des colorants spécifiques tels que le DAPI, le FITC, le TRITC et le Texas Red.